
Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome

 , ,
, ,  and
and
Abstract
1. Introduction
2. Cell Membrane Fluidity and Permeability
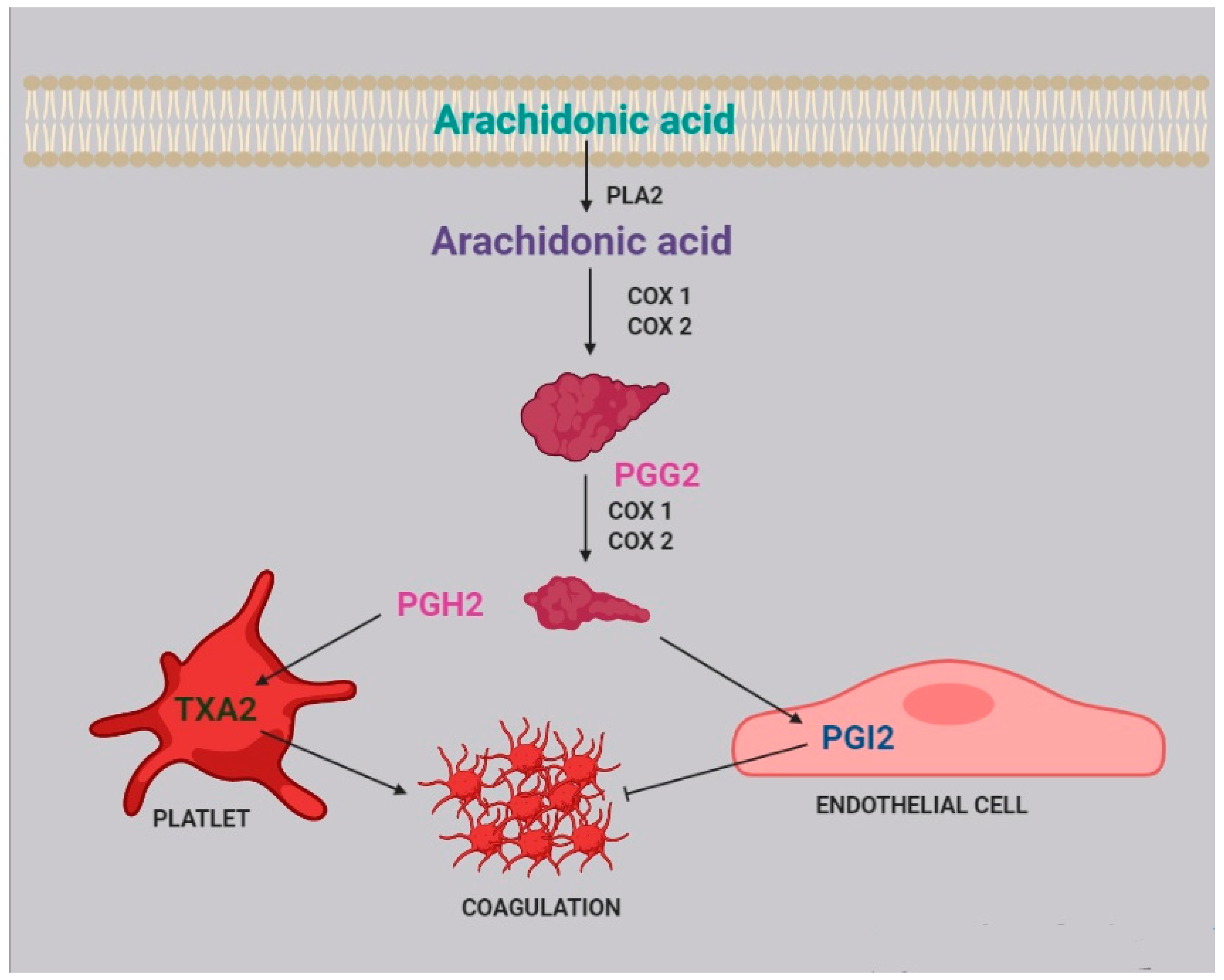
3. Platelet Aggregation and Coagulation
4. Immune System
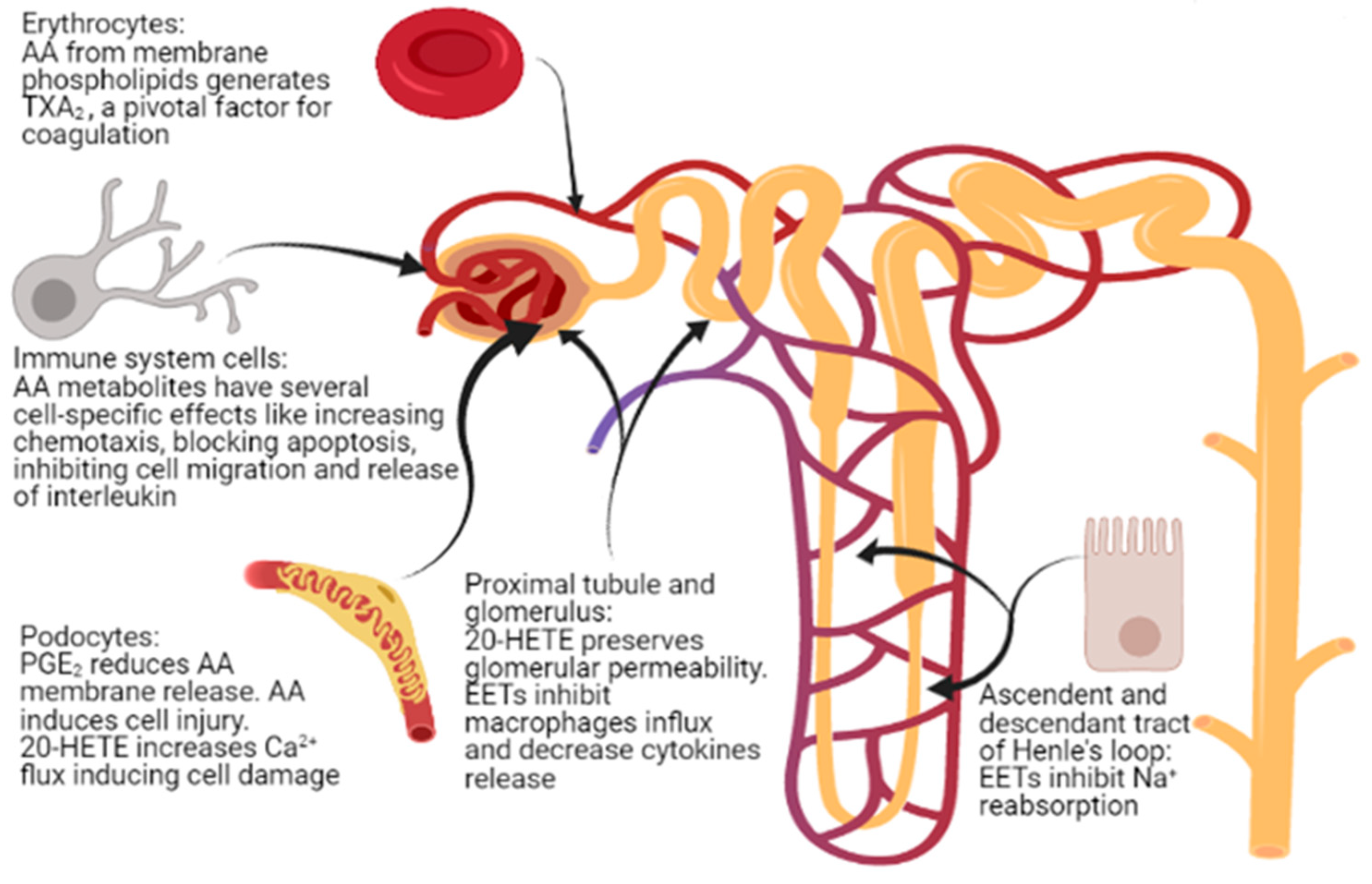
5. Kidney Glomerular and Tubular Function
6. Podocyte Physiopathology and Infections
7. Renal Fibrosis
8. Drug and Gene Interactions
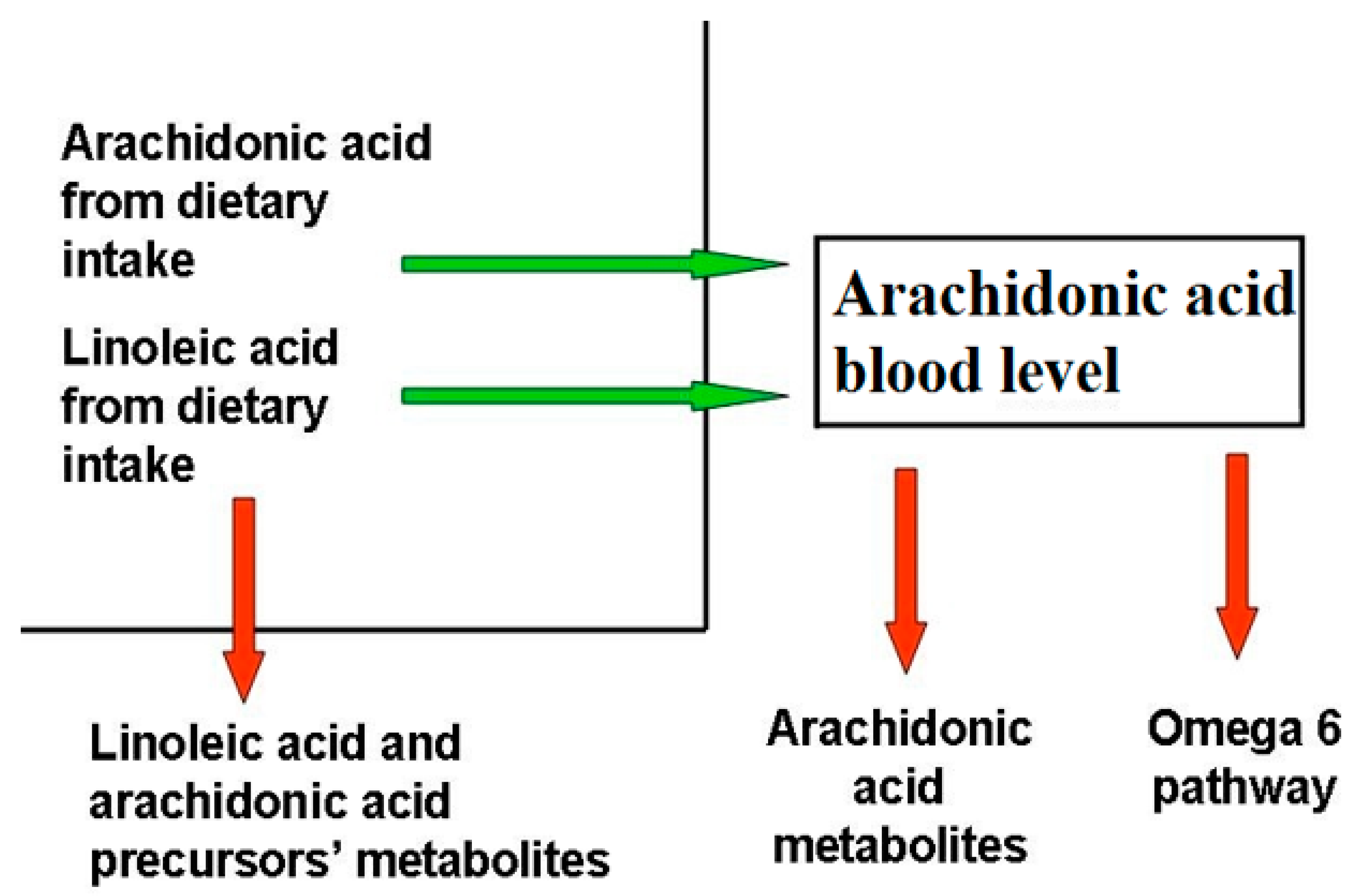
9. Dietary Balance Between AA and LA and AA Sources
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Noone, D.G.; Iijima, K.; Parekh, R. Idiopathic Nephrotic Syndrome in Children. Lancet 2018, 392, 61–74. [Google Scholar] [CrossRef]
- Teoh, C.W.; Robinson, L.A.; Noone, D. Perspectives on edema in childhood nephrotic syndrome. Am. J. Physiol. Ren. Physiol. 2015, 309, F575–F582. [Google Scholar] [CrossRef]
- Dorhout Mees, E.J.; Koomans, H.A. Understanding the nephrotic syndrome: What’s new in a decade? Nephron 1995, 70, 1–10. [Google Scholar] [CrossRef]
- Eneman, B.; Levtchenko, E.; van den Heuvel, B.; Van Geet, C.; Freson, K. Platelet abnormalities in nephrotic syndrome. Pediatr. Nephrol. 2016, 31, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, M.; Massella, L.; Ruggiero, B.; Emma, F. Minimal Change Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S. Pathology of Podocytopathies Causing Nephrotic Syndrome in Children. Front. Pediatr. 2016, 4, 32. [Google Scholar] [CrossRef]
- Eddy, A.A.; Symons, J.M. Nephrotic Syndrome in Childhood. Lancet 2003, 362, 629–639. [Google Scholar] [CrossRef]
- Risé, P.; Tragni, E.; Ghezzi, S.; Agostoni, C.; Marangoni, F.; Poli, A.; Catapano, A.L.; Siani, A.; Iacoviello, L.; Galli, C. Different patterns characterize Omega 6 and Omega 3 long chain polyunsaturated fatty acid levels in blood from Italian infants, children, adults and elderly. Prostagland. Leukot. Essent. Fat. Acids 2013, 89, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Cunnnane, S.C.; Kent, E.T.; McAdoo, K.R.; Caldwell, D.; Lin, A.N.; Carter, D.M. Abnormalities of Plasma and Erythrocyte Essential Fatty Acid Composition in Epidermolysis Bullosa: Influence of Treatment with Diphenylhydantoin. J. Investig. Dermatol. 1987, 89, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. Prostagland. Leukot. Essent. Fat. Acids 2010, 83, 121–129. [Google Scholar] [CrossRef]
- Cheng, S.; Chen, Y.L. Study on erythrocyte membrane fluidity by laser Raman spectroscopy. Cell Biol. Int. Rep. 1988, 12, 205–211. [Google Scholar] [PubMed]
- Pompéia, C.; Lopes, L.R.; Miyasaka, C.K.; Procópio, J.; Sannomiya, P.; Curi, R. Effect of fatty acids on leukocyte function. Braz. J. Med. Biol. Res. 2000, 33, 1255–1268. [Google Scholar] [CrossRef]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.H.; Nascimento, K.S.; Freire, M.M.; Moreira, O.C.; Scofano, H.M.; Barrabin, H.; Mignaco, J.A. Mechanism of modulation of the plasma membrane Ca(2+)-ATPase by arachidonic acid. Prostagland. Other Lipid Mediat. 2008, 87, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef]
- Meves, H. Arachidonic acid and ion channels: An update. Br. J. Pharmacol. 2008, 155, 4–16. [Google Scholar] [CrossRef]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef]
- Van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef]
- Beck, R.; Bertolino, S.; Abbot, S.E.; Aaronson, P.I.; Smirnov, S.V. Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ. Res. 1998, 83, 923–931. [Google Scholar] [CrossRef]
- Nelson, G.J.; Schmidt, P.C.; Bartolini, G.; Kelley, D.S.; Kyle, D. The effect of dietary arachidonic acid on platelet function, platelet fatty acid composition, and blood coagulation in humans. Lipids 1997, 32, 421–425. [Google Scholar] [CrossRef]
- Trostchansky, A.; Moore-Carrasco, R.; Fuentes, E. Oxydative pathways of arachidonic acid as targets for regulation of platelet activation. Prostaglandines Other Lipid Mediat. 2019, 15, 106382. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef]
- Rasedee, A.; Feldman, B.F. Nephrotic syndrome: A platelet hyperaggregability state. Vet. Res. Commun. 1985, 9, 199–211. [Google Scholar] [CrossRef]
- Davì, G.; Averna, M.; Catalano, I.; Barbagallo, C.; Ganci, A.; Notarbartolo, A.; Ciabattoni, G.; Patrono, C. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation 1992, 85, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Carsetti, R.; Cascioli, S.; Serafinelli, J.; Emma, F.; Vivarelli, M. B cell phenotype in pediatric idiopathic nephrotic syndrome. Pediatr. Nephrol. 2019, 34, 177–181. [Google Scholar] [CrossRef]
- Chebotareva, N.; Bobkova, I.; Lysenko, L. T regulatory cells in renal tissue of patients with nephrotic syndrome. Pediatr. Int. 2020, 62, 884–885. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.T.; Gbadegesin, R.A. Aberrant IgM on T cells: Biomarker or pathogenic factor in childhood nephrotic syndrome? Kidney Int. 2019, 96, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, F.T.L.; Melo, G.E.B.A.; Cordeiro, T.M.; Feracin, V.; Vieira, E.R.; Pereira, W.F.; Pinheiro, S.V.B.; Miranda, A.S.; Simões-E-Silva, A.C. T-lymphocyte-expressing inflammatory cytokines underlie persistence of proteinuria in children with idiopathic nephrotic syndrome. J. Pediatr. 2018, 94, 546–553. [Google Scholar] [CrossRef]
- Alsharidah, A.S.; Alzogaibi, M.A.; Bayoumy, N.M.; Alghonaim, M. Neutrophil chemokines levels in different stages of nephrotic syndrome. Saudi J. Kidney Dis. Transplant. 2017, 28, 1256–1263. [Google Scholar] [CrossRef]
- Calder, P.C. The relationship between the fatty acid composition of immune cells and their function. Prostagland. Leukot. Essent. Fat. Acids 2008, 79, 101–108. [Google Scholar] [CrossRef]
- Triggiani, M.; Oriente, A.; de Crescenzo, G.; Rossi, G.; Marone, G. Biochemical functions of a pool of arachidonic acid associated with triglycerides in human inflammatory cells. Int. Arch. Allergy Immunol. 1995, 107, 261–263. [Google Scholar] [CrossRef]
- Wei, J.; Gronert, K. Eicosanoid and specialized preresolving mediator regulation of lymphoid cells. Trends Biochem. Sci. 2019, 44, 214–225. [Google Scholar] [CrossRef]
- Hanna, V.S.; Hafez, E.A. Synopsis of arachidonic acid metabolism—A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fu, X.; Chen, Q.; Part, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic acid metabolism and kidney inflammation. Int. J. Mol. Sci. 2019, 20, 3683. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar] [CrossRef]
- Legler, D.F.; Bruckner, M.; Uetz-von Allmen, E.; Krause, P. Prostaglandin E2 at new glance: Novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 2010, 42, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Harizi, H.; Gualde, N. The impact of eicosanoids on the crosstalk between innate and adaptive immunity: The key roles of dendritic cells. Tissue Antigens 2005, 65, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, C.; Thomas, D.W.; Tilley, S.L.; Nguyen, M.T.; Mannon, R.; Koller, B.H.; Coffman, T.M. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J. Clin. Investig. 2001, 108, 1229–1235. [Google Scholar] [CrossRef]
- Roper, R.L.; Phipps, R.P. Prostaglandin E2 and cAMP inhibit B lymphocyte activation and simultaneously promote IgE and IgG1 synthesis. J. Immunol. 1992, 149, 2984–2991. [Google Scholar]
- Goodarzi, K.; Goodarzi, M.; Tager, A.M.; Luster, A.D.; von Andrian, U.H. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 2003, 4, 965–973. [Google Scholar] [CrossRef]
- Yang, L.; Mäki-Petäjä, K.; Cheriyan, J.; McEniery, C.; Wilkinson, I.B. The role of epoxyeicosatrienoic acids in the cardiovascular system. Br. J. Clin. Pharmacol. 2015, 80, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Chen, G.Z.; Wang, Y.; Wang, D.W. Role of cytochrome P450 epoxygenase-dependent arachidonic acid metabolites in kidney physiology and diseases. Sheng Li Xue Bao 2018, 70, 591–599. [Google Scholar] [PubMed]
- Wang, D.; DuBois, R.N. Epoxyeicosatrienoic acids: A double-edged sword in cardiovascular diseases and cancer. J. Clin. Investig. 2012, 122, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Vascular cytochrome p450 enzymes: Physiology and pathophysiology. Trends Cardiovasc. Med. 2008, 18, 20–25. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, S.P.; Toews, M.L.; Imig, J.D.; Hwang, S.H.; Hammock, B.D.; Padanilam, B.J. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 308, F131–F139. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef]
- Carroll, M.A.; Cheng, M.K.; Liclican, E.L.; Li, J.; Doumad, A.B.; McGiff, J.C. Purinoceptors in renal microvessels: Adenosine-activated and cytochrome P450 monooxygenase-derived arachidonate metabolites. Pharmacol. Rep. 2005, 57, 191–195. [Google Scholar]
- Williams, J.M.; Sharma, M.; Anjaiahh, S.; Falck, J.R.; Roman, R.J. Role of endogenous CYP450 metabolites of arachidonic acid in maintaining the glomerular protein permeability barrier. Am. J. Physiol. Ren. Physiol. 2007, 293, F501–F505. [Google Scholar] [CrossRef]
- Fan, F.; Roman, R.J. Effect of Cytochrome P450 Metabolites of Arachidonic Acid in Nephrology. J. Am. Soc. Nephrol. 2017, 28, 2845–2855. [Google Scholar] [CrossRef]
- Nowicki, S.; Chen, S.L.; Aizman, O.; Cheng, X.J.; Li, D.; Nowicki, C.; Nairn, A.; Greengard, P.; Aperia, A. 20-Hydroxyeicosa-tetraenoic acid (20 HETE) activates protein kinase C. Role in regulation of rat renal Na+,K+-ATPase. J. Clin. Investig. 1997, 99, 1224–1230. [Google Scholar] [CrossRef]
- Roshanravan, H.; Kim, E.Y.; Dryer, S.E. 20-Hydroxyeicosatetraenoic Acid (20-HETE) Modulates in Podocytes. Front. Physiol. 2016, 7, 351. [Google Scholar] [CrossRef]
- Shatat, I.F.; Becton, L.J.; Woroniecki, R.P. Hypertension in childhood nephrotic syndrome. Front. Pediatr. 2019, 7, 287. [Google Scholar] [CrossRef]
- Quigley, R.; Baum, M.; Reddy, K.M.; Griener, J.C.; Falck, J.R. Effects of 20-HETE and 19(s)-HETE on rabbit proximal straight tubule volume transport. Am. J. Physiol. Ren. Physiol. 2000, 278, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Osama, H.E.; Sheriff, M.S.; Anwar, M.; Ayman, O.S. Clinical implication of 20-hydroeicosatetraenoic acid in the kidney, liver, lung and brain. An emerging therapeutic target. Pharmaceutics 2017, 9, 9. [Google Scholar]
- Han, H.; Wang, S.; Liang, Y.; Lin, J.; Shi, L.; Ye, L.; Song, S.; He, M.; Li, S.; Chen, F.; et al. Respiratory tract infection: A risk factor for the onset and relapse of adult onset minimal changes disease in Southern China. BioMed Res. Int. 2018, 2018, 1657208. [Google Scholar] [CrossRef]
- Mishra, O.P.; Abhinay, A.; Mishra, R.N.; Prasad, R.; Pohl, M. Can we predict relapses in children with idiopathic steroid-sensitive nephrotic syndrome? J. Trop. Pediatr. 2013, 59, 343–349. [Google Scholar] [CrossRef]
- Manta, M.; Singh, S. Infection associated relapses in children with nephrotic syndrome: A short-term outcome study. Saudi J. Kidney Dis. Transplant. 2019, 30, 1245–1253. [Google Scholar] [CrossRef]
- Ha, V.T.; Lainšček, D.; Gesslbauer, B.; Jarc-Jovičić, E.; Hyötyläinen, T.; Ilc, N.; Lakota, K.; Tomšič, M.; van de Loo, F.A.J.; Bochkov, V.; et al. Synergy between 15-lipoxygenase and secreted PLA2 promotes inflammation by formation of TLR4 agonists from extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 25679–25689. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.S.; Zhao, X.; Inscho, E.W.; Imig, J.D. 12-Hydroxyeicosatetraenoic acid participates in angiotensin II afferent arteriolar vasoconstriction by activating L-type calcium channels. J. Lipid Res. 2003, 44, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Andreani, M.; Olivier, J.L.; Berenbaum, F.; Raymondjean, M.; Béréziat, G. Transcriptional regulation of inflammatory secreted phospholipases A(2). Biochim. Biophys. Acta 2000, 1488, 149–158. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, M.; Chen, L.; Zhang, W.; Huang, Y.; Luo, H.; Li, L.; He, H. The anti-inflammatory effects of Yunnan Baiyao are involved in regulation of the phospholipase A2/arachidonic acid metabolites pathways in acute inflammation rat model. Mol. Med. Rep. 2017, 16, 4045–4053. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, L.I.; Rahal, S.S.; Kennedy, C.R. PGE2 reduces arachidonic acid release in murine podocytes: Evidence for an autocrine feedback loop. Am. J. Physiol. Cell Physiol. 2003, 284, C302–C309. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pan, Y.; Wu, Y.; Lin, S.; Dai, B.; Chen, H.; Wan, J. Excessive arachidonic acid induced actin bunching remodeling and podocyte injury via a PKA-c-Abl dependent pathway. Exp. Cell Res. 2020, 388, 111808. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wan, J.; Liu, Y.; Yang, Q.; Liang, W.; Singhal, P.C.; Saleem, M.A.; Ding, G. sPLA2 IB induces human podocyte apoptosis via the M-type phospholipase A2 receptor. Sci. Rep. 2014, 4, 6660. [Google Scholar] [CrossRef]
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36. [Google Scholar] [CrossRef]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef]
- Priante, G.; Musacchio, E.; Valvason, C.; Baggio, B. EPA and DHA suppress AngII- and arachidonic acid-induced expression of profibrotic genes in human mesangial cells. J. Nephrol. 2009, 22, 137–143. [Google Scholar]
- Zimpelmann, J.; Burns, K.D. Angiotensin-(1-7) activates growth-stimulatory pathways in human mesangial cells. Am. J. Physiol. Ren. Physiol. 2009, 296, F337–F346. [Google Scholar] [CrossRef]
- Zhang, C.; Booz, G.W.; Yu, Q.; He, X.; Wang, S.; Fan, F. Conflicting roles of 20-HETE in hypertension and renal end organ damage. Eur. J. Pharmacol. 2018, 833, 190–200. [Google Scholar] [CrossRef]
- Cheng, J.; Garcia, V.; Ding, Y.; Wu, C.C.; Thakar, K.; Falck, J.R.; Ramu, E.; Schwartzman, M.L. Induction of angiotensin-converting enzyme and activation of the renin-angiotensin system contribute to 20-hydroxyeicosatetraenoic acid-mediated endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1917–1924. [Google Scholar] [CrossRef]
- Balakumar, P.; Sambathkumar, R.; Mahadevan, N.; Muhsinah, A.B.; Alsayari, A.; Venkateswaramurthy, N.; Jagadeesh, G. A potential role of the renin-angiotensin-aldosterone system in epithelial-to-mesenchymal transition-induced renal abnormalities: Mechanisms and therapeutic implications. Pharmacol. Res. 2019, 146, 104314. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H. Prostaglandin E2 Induces Skin Aging via E-Prostanoid 1 in Normal Human Dermal Fibroblasts. Int. J. Mol. Sci. 2019, 20, 5555. [Google Scholar] [CrossRef] [PubMed]
- Lausada, N.; de Gómez Dumm, I.N.; Raimondi, J.C.; de Alaniz, M.J. Effect of cyclosporine and sirolimus on fatty acid desaturase activities in cultured HEPG2 cells. Transplant. Proc. 2009, 41, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Whisler, R.L.; Lindsey, J.A.; Proctor, K.V.; Morisaki, N.; Cornwell, D.G. Characteristics of cyclosporine induction of increased prostaglandin levels from human peripheral blood monocytes. Transplantation 1984, 38, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Bukhave, K.; Ahnfelt-Rønne, I.; Elmgreen, J. Arachidonic acid metabolism in human neutrophils: Lack of effect of cyclosporine A. Int. J. Immunopharmacol. 1986, 8, 419–426. [Google Scholar] [CrossRef]
- Sipka, S.; Szücs, K.; Szántó, S.; Kovács, I.; Lakos, G.; Antal-Szalmás, P.; Szegedi, G.; Gergely, P. Inhibition of calcineurin activity and protection against cyclosporine A induced cytotoxicity by prednisolone sodium succinate in human peripheral mononuclear cells. Immunopharmacology 2000, 48, 87–92. [Google Scholar] [CrossRef]
- Wondimu, B.; Modéer, T. Cyclosporin A upregulates prostaglandin E2 production in human gingival fibroblasts challenged with tumor necrosis factor alpha in vitro. J. Oral Pathol. Med. 1997, 26, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.E.; Jones, J.P., 3rd; Kalhorn, T.F.; Farin, F.M.; Stapleton, P.L.; Davis, C.L.; Perkins, J.D.; Blough, D.K.; Hebert, M.F.; Thummel, K.E.; et al. Role of cytochrome P450 2C8 and 2J2 genotypes in calcineurin inhibitor-induced chronic kidney disease. Pharmacogenet. Genom. 2008, 18, 943–953. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Paul-Clark, M.; Van Hal, P.T. Differential modulation of glucocorticoid action by FK506 in A549 cells. Biochem. J. 2003, 376 Pt 1, 285–290. [Google Scholar] [CrossRef]
- Mariee, A.D.; Abd-Ellah, M.F. Protective effect of docosahexaenoic acid against cyclosporine A-induced nephrotoxicity in rats: A possible mechanism of action. Ren. Fail. 2011, 33, 66–71. [Google Scholar] [CrossRef]
- Hirunpanich, V.; Sato, H. Improvement of cyclosporine A bioavailability by incorporating ethyl docosahexaenoate in the microemulsion as an oil excipient. Eur. J. Pharm. Biopharm. 2009, 73, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, A.; Agostoni, C. Long-Chain ω-3 Polyunsaturated Fatty Acids: Do Genetic Steps Match Metabolic Needs? J. Nutr. 2019, 149, 1690–1691. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. Evolutionary aspects of diet: The omega-6/omega-3 ratio and the brain. Mol. Neurobiol. 2011, 44, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Nara, T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004, 24, 345–376. [Google Scholar] [CrossRef]
- Ameur, S.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.V.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty acid metabolism: A human specific haplotype increasing the biosyntesis of long-chain omega-3 and omega -6 fatty acid. Am. J. Hum. Genet. 2012, 90, 809–820. [Google Scholar] [CrossRef]
- Marangoni, F.; Agostoni, C.; Borghi, C.; Catapano, A.L.; Cena, H.; Ghiselli, A.; La Vecchia, C.; Lercker, G.; Manzato, E.; Pirillo, A.; et al. Dietary linoleic acid and human health: Focus on cardiovascular and cardiometabolic effects. Atherosclerosis 2020, 292, 90–98. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Zamora, D.; Makriyannis, A.; Wood, J.T.; Mann, J.D.; Faurot, K.R.; MacIntosh, B.A.; Majchrzak-Hong, S.F.; Gross, J.R.; Courville, A.B.; et al. Diet-induced changes in n-3- and n-6-derived endocannabinoids and reductions in headache pain and psychological distress. J. Pain 2015, 16, 707–716. [Google Scholar] [CrossRef]
- Forsyth, S.; Gautier, S.; Salem, N., Jr. Global Estimates of Dietary Intake of Docosahexaenoic Acid and Arachidonic Acid in Developing and Developed Countries. Ann. Nutr. Metab. 2016, 68, 258–267. [Google Scholar] [CrossRef]
- US National Institutes of Health. Food Sources of Arachidonic Acid; US National Institutes of Health: Bethesda, MD, USA, 2005.
- Li, D.; Ng, A.; Mann, N.J.; Sinclair, A.J. Contribution of meat fat to dietary arachidonic acid. Lipids 1998, 33, 437–440. [Google Scholar] [CrossRef]
- Abedi, E.; Sahari, M.A. Long-chain polyunsaturated fatty acid sources and evaluation of their nutritional and functional properties. Food Sci. Nutr. 2014, 2, 443–463. [Google Scholar] [CrossRef]
- Shanab, S.M.M.; Hafez, R.M.; Fouad, A.S. A review on algae and plants as potential source of arachidonic acid. J. Adv. Res. 2018, 11, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Neonates | Children | Adults | Elderly | |
|---|---|---|---|---|
| Linoleic acid (%) | 4.61 ± 1.06 | 17.67 ± 1.92 | 18.41 ± 2.87 | 17.64 ± 2.89 |
| Arachidonic acid (%) | 13.14 ± 1.73 | 8.33 ± 1.04 | 8.51 ± 1.38 | 8.32 ± 1.40 |
| Total omega 6 (%) | 22.99 ± 2.13 | 28.97 ± 2.19 | 29.79 ± 3.13 | 28.78 ± 3.24 |
| Total saturated fatty acids (%) | 46.10 ± 3.16 | 44.32 ± 1.61 | 39.47 ± 2.3 | 39.83 ± 2.16 |
| Total monounsaturated fatty acids (%) | 26.15 ± 2.76 | 24.39 ± 2.07 | 27.20 ± 3.08 | 27.83 ± 3.27 |
| Total omega 3 (%) | 4.76 ± 0.89 | 2.31 ± 0.50 | 3.54 ± 1.05 | 3.55 ± 0.95 |
| Cell Type | AA Metabolite | Effect |
|---|---|---|
| Basophil | PGD2 | Stimulates basophil chemotaxis |
| Eosinophil | PGD2 | Stimulates eosinophil chemotaxis |
| Blocks eosinophil apoptosis | ||
| Activates eosinophils | ||
| Naive t cell | TXA2 | Inhibits proliferation of naive T cells |
| B-cell | PGE2 | Enhances IgE class switching by B cells |
| Dendritic cell | LTB4 | Stimulates DC production of IL-6 |
| LTC4 | Participates in cell migration | |
| Enhances cells activation and functions | ||
| PGD2 | Inhibits cells migration | |
| PGE2 | Stimulates IL-10 production | |
| Modulates cell migration | ||
| Downregulates major histocompatibility complex C class II expression | ||
| Inhibits IL-12 and IFN-ã production | ||
| Inhibits the expression of CCL3/CCL4 | ||
| PGJ2 | Induces apoptosis | |
| TXA2 | Inhibits interaction with T cell | |
| Langerhans cell | PGE2 | Promotes the migration and maturation of Langerhans cells |
| PGD2 | Inhibits cells migration | |
| Lymphocyte | PGE2 | Inhibits interactions with endothelial cell |
| Macrophage | PGE2 | Suppresses cytokine production |
| Suppresses chemokine expression | ||
| PGJ2 | Inhibits release of IL-10 and IL-12 | |
| Mast cell | PGE2 | Enhances antigen-stimulated degranulation |
| Neutrophil | LTB4 | Activates cells |
| NK cell | PGE2 | Inhibits IL-12 and IFN-ã production |
| T cell | LTB4 | Enhances cell recruitment |
| PGE2 | Inhibits cell proliferation | |
| TXA2 | Inhibits interactions with dendritic cells | |
| Regulates the elimination of self-reactive cells | ||
| Increases cell proliferation and activation | ||
| Enhances local cytotoxic cell function | ||
| Th2 | PGD2 | Stimulates chemotaxis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turolo, S.; Edefonti, A.; Mazzocchi, A.; Syren, M.L.; Morello, W.; Agostoni, C.; Montini, G. Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome. Int. J. Mol. Sci. 2021, 22, 5452. https://doi.org/10.3390/ijms22115452
Turolo S, Edefonti A, Mazzocchi A, Syren ML, Morello W, Agostoni C, Montini G. Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome. International Journal of Molecular Sciences. 2021; 22(11):5452. https://doi.org/10.3390/ijms22115452
Chicago/Turabian StyleTurolo, Stefano, Alberto Edefonti, Alessandra Mazzocchi, Marie Louise Syren, William Morello, Carlo Agostoni, and Giovanni Montini. 2021. "Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome" International Journal of Molecular Sciences 22, no. 11: 5452. https://doi.org/10.3390/ijms22115452
APA StyleTurolo, S., Edefonti, A., Mazzocchi, A., Syren, M. L., Morello, W., Agostoni, C., & Montini, G. (2021). Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome. International Journal of Molecular Sciences, 22(11), 5452. https://doi.org/10.3390/ijms22115452