
Lemur Tyrosine Kinases and Prostate Cancer: A Literature Review




Abstract
1. Introduction
Prostate Cancer
2. LMTK Proteins
2.1. LMTKs and Cancer
2.2. Catalytic Specificity of LMTKs
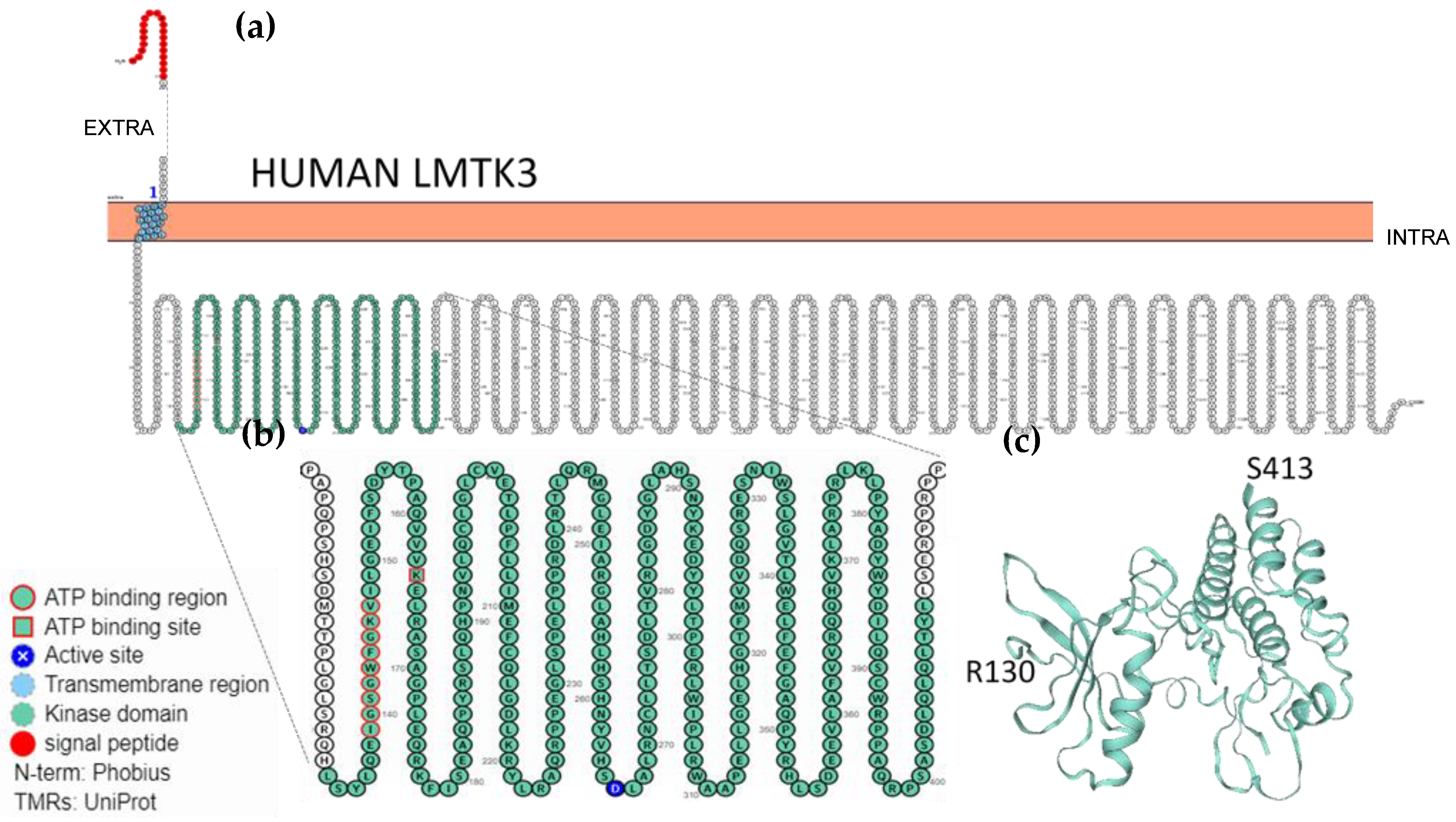
2.3. Localisation, Membrane Topology and Structural Features of LMTKs
3. Literature Review
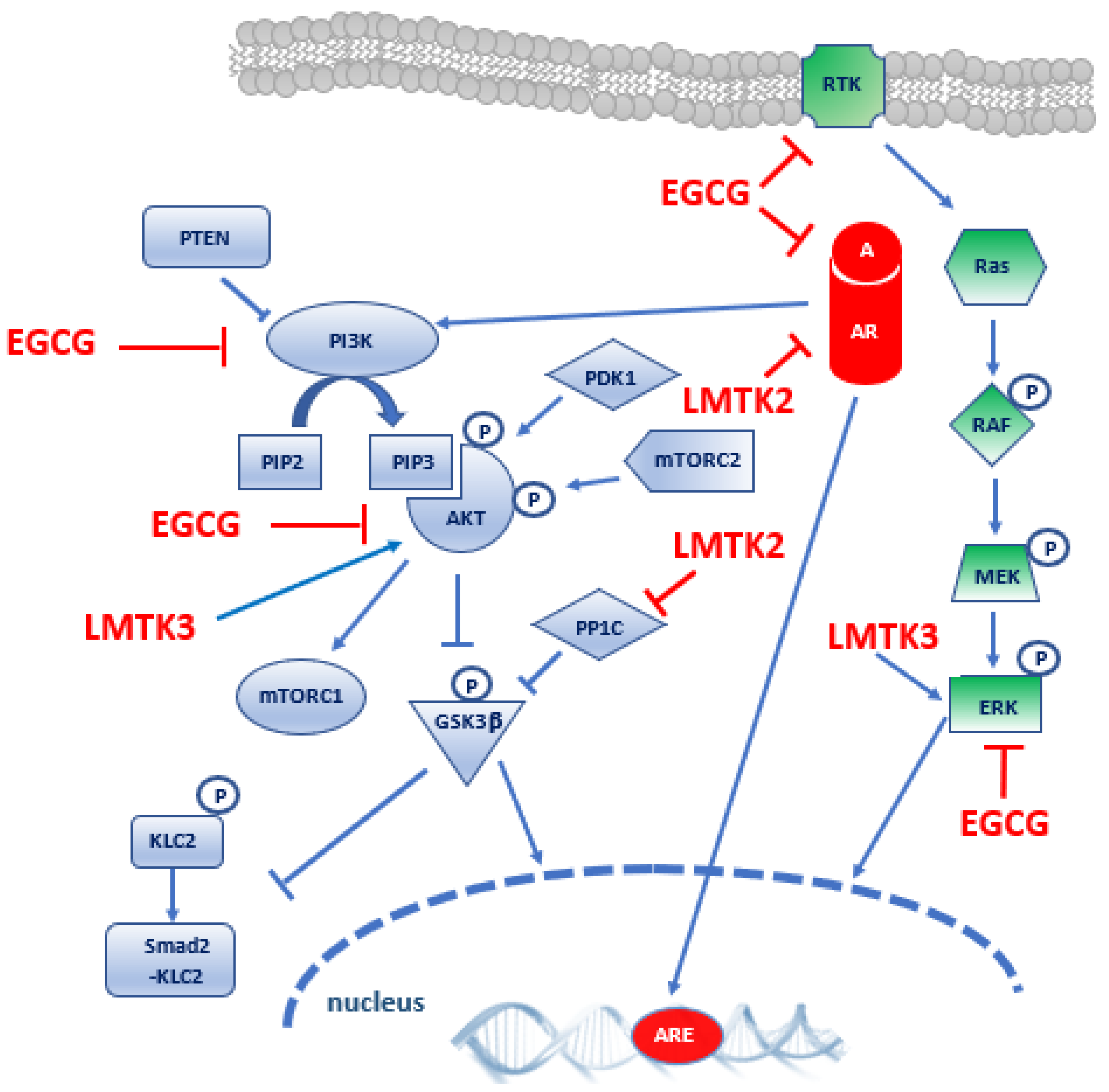
3.1. Evidence for LMTK2 Involvement in Prostate Cancer
3.2. Evidence for LMTK3 Involvement in Prostate Cancer
4. Epigallocatechin-3-Gallate
5. Discussion
6. Methods: Search Strategy and Study Selection
Author Contributions
Funding
Conflicts of Interest
References
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L. Lo The crucial role of protein phosphorylation in cell signalingand its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Wilson, L.J.; Linley, A.; Hammond, D.E.; Hood, F.E.; Coulson, J.M.; MacEwan, D.J.; Ross, S.J.; Slupsky, J.R.; Smith, P.D.; Eyers, P.A.; et al. New Perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018, 78, 15–29. [Google Scholar] [CrossRef]
- Bradley, D.; Beltrao, P. Evolution of protein kinase substrate recognition at the active site. PLoS Biol. 2019, 17, e3000341. [Google Scholar] [CrossRef]
- Hanks, S.K. Genomic analysis of the eukaryotic protein kinase superfamily: A perspective. Genome Biol. 2003, 4, 111. [Google Scholar] [CrossRef][Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor tyrosine kinase-targeted cancer therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; Román-Gil, M.S.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine kinase receptors in oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharm. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, T.; Zhang, J. Natural tyrosine kinase inhibitors acting on the epidermal growth factor receptor: Their relevance for cancer therapy. Pharm. Res. 2020, 161, 105164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bilal, M.; Raza, A.; Khan, M.I.; Mehmood, S.; Hayat, U.; Hassan, S.T.S.; Iqbal, H.M.N. Tyrosine kinase inhibitors and their unique therapeutic potentialities to combat cancer. Int. J. Biol. Macromol. 2021, 168, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Smit, E.F.; Groen, H.J.M.; Horn, L.; Gettinger, S.; Camidge, D.R.; Riely, G.J.; Wang, B.; Fu, Y.; Chand, V.K.; et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014, 4, 1036–1045. [Google Scholar] [CrossRef]
- NIH-Illuminating the Druggable Genome (IDG). Available online: https://commonfund.nih.gov/IDG (accessed on 25 March 2021).
- Oprea, T.I. Exploring the dark genome: Implications for precision medicine. Mamm. Genome 2019, 30, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Wendler, F. The LMTK-family of kinases: Emerging important players in cell physiology and disease pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.; Kote-Jarai, Z.; Giles, G.G.; Al Olama, A.A.; Jugurnauth, S.K.; Mulholland, S.; Leongamornlert, D.A.; Edwards, S.M.; Morrison, J.; Field, H.I.; et al. Identification of new genetic risk factors for prostate cancer. Asian J. Androl. 2009, 11, 49–55. [Google Scholar] [CrossRef]
- Kohaar, I.; Petrovics, G.; Srivastava, S. A rich array of prostate cancer molecular biomarkers: Opportunities and challenges. Int. J. Mol. Sci. 2019, 20, 1813. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, D.; Spring, D.J.; Depinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef]
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef]
- Baade, P.D.; Youlden, D.R.; Krnjacki, L.J. International epidemiology of prostate cancer: Geographical distribution and secular trends. Mol. Nutr. Food Res. 2009, 53, 171–184. [Google Scholar] [CrossRef]
- Kheirandish, P.; Chinegwundoh, F. Ethnic differences in prostate cancer. Br. J. Cancer 2011, 105, 481–485. [Google Scholar] [CrossRef]
- Taitt, H.E. Global Trends and Prostate Cancer: A Review of Incidence, Detection, and Mortality as Influenced by Race, Ethnicity, and Geographic Location. Am. J. Mens. Health 2018, 12, 1807–1823. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Reig, Ò.; Marín-Aguilera, M.; Carrera, G.; Jiménez, N.; Paré, L.; García-Recio, S.; Gaba, L.; Pereira, M.V.; Fernández, P.; Prat, A.; et al. TMPRSS2-ERG in Blood and Docetaxel Resistance in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 70, 709–713. [Google Scholar] [CrossRef]
- Beltran, H.; Romanel, A.; Conteduca, V.; Casiraghi, N.; Sigouros, M.; Franceschini, G.M.; Orlando, F.; Fedrizzi, T.; Ku, S.Y.; Dann, E.; et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J. Clin. Investig. 2020, 130, 1653–1668. [Google Scholar] [CrossRef]
- Li, Y.K.; Moughan, J.; Al-Saleem, T.; Hammond, E.H.; Venkatesan, V.; Rosenthal, S.A.; Ritter, M.A.; Sandler, H.M.; Hanks, G.E.; Shipley, W.U.; et al. Bcl-2 and bax expression predict prostate cancer outcome in men treated with androgen deprivation and radiotherapy on radiation therapy oncology group protocol 92-02. Clin. Cancer Res. 2007, 13, 3585–3590. [Google Scholar] [CrossRef]
- Yadav, S.; Anbalagan, M.; Baddoo, M.; Chellamuthu, V.K.; Mukhopadhyay, S.; Woods, C.; Jiang, W.; Moroz, K.; Flemington, E.K.; Makridakis, N. Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 2020, 39, 4299–4311. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.H.; Feddersen, S.; Albitar, M.; Poulsen, C.A.; Lund, M.; Pedersen, T.B.; Mortensen, M.A.; Lund, L. A prospective study of a urine and plasma biomarker test for the prediction of gleason ≥3 + 4 prostate cancer in a mixed cohort. Scand. J. Urol. 2020, 54, 323–327. [Google Scholar] [CrossRef]
- Aslan, R.; Alp, H.H.; Eryılmaz, R.; Huyut, Z.; Sevim, M.; Araz, S.; Ertas, K.; Taken, K. Can the Irisin be a Biomarker for Prostate Cancer? A Case Control Study. Asian Pac. J. Cancer Prev. 2020, 21, 505–509. [Google Scholar] [CrossRef]
- Liu, Z.; Zhong, J.; Cai, C.; Lu, J.; Wu, W.; Zeng, G. Immune-related biomarker risk score predicts prognosis in prostate cancer. Aging 2020, 12, 22776–22789. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Liang, J.; Hu, J.; Mi, Y.; Ruan, J.; Zhang, J.; Wang, Z.; Hu, Q.; Jiang, H.; Ding, Q. TRAF2 is a valuable prognostic biomarker in patients with prostate cancer. Med. Sci. Monit. 2017, 23, 4192–4204. [Google Scholar] [CrossRef]
- Govers, T.M.; Caba, L.; Resnick, M.J. Cost-Effectiveness of Urinary Biomarker Panel in Prostate Cancer Risk Assessment. J. Urol. 2018, 200, 1221–1226. [Google Scholar] [CrossRef]
- Huang, Y.; Pledgie, A.; Casero, R.A.; Davidson, N.E. Molecular mechanisms of polyamine analogs in cancer cells. Anticancer. Drugs 2005, 16, 229–241. [Google Scholar] [CrossRef]
- Ramberg, H.; Grytli, H.H.; Nygård, S.; Wang, W.; Ögren, O.; Zhao, S.; Løvf, M.; Katz, B.; Skotheim, R.I.; Bjartell, A.; et al. PBX3 is a putative biomarker of aggressive prostate cancer. Int. J. Cancer 2016, 139, 1810–1820. [Google Scholar] [CrossRef]
- Cai, B.; Peng, J.H. Increased expression of miR-494 in serum of patients with prostate cancer and its potential diagnostic value. Clin. Lab. 2019, 65, 1507–1512. [Google Scholar] [CrossRef]
- Ikeda, S.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Next-generation sequencing of prostate cancer: Genomic and pathway alterations, potential actionability patterns, and relative rate of use of clinical-grade testing. Cancer Biol. Ther. 2019, 20, 219–226. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Investig. 2018, 128, 4441–4453. [Google Scholar] [CrossRef]
- Bishop, M.R.; Huskey, A.L.W.; Hetzel, J.; Merner, N.D. A research-based gene panel to investigate breast, ovarian and prostate cancer genetic risk. PLoS ONE 2019, 14, e0220929. [Google Scholar] [CrossRef]
- Cruz, D.F.; Farinha, C.M.; Swiatecka-Urban, A. Unraveling the function of lemur tyrosine kinase 2 network. Front. Pharm. 2019, 10, 24. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; Ji, M.; Shi, L.; Xu, B.; Jiang, J.; Wu, C. Prognostic role of lemur tyrosine kinase 3 in postoperative gastric cancer. Mol. Clin. Oncol. 2014, 2, 756–760. [Google Scholar] [CrossRef]
- Jiang, T.; Lu, X.; Yang, F.; Wang, M.; Yang, H.; Xing, N. LMTK3 promotes tumorigenesis in bladder cancer via the ERK/MAPK pathway. FEBS Open Bio 2020, 10, 2107–2121. [Google Scholar] [CrossRef]
- Conti, A.; Majorini, M.T.; Fontanella, E.; Bardelli, A.; Giacca, M.; Delia, D.; Mano, M.; Lecis, D. Lemur tyrosine kinase 2 (LMTK2) is a determinant of cell sensitivity to apoptosis by regulating the levels of the BCL2 family members. Cancer Lett. 2017, 389, 59–69. [Google Scholar] [CrossRef]
- Zhang, R.; Li, X.; Wei, L.; Qin, Y.; Fang, J. Lemur tyrosine kinase 2 acts as a positive regulator of NF-κB activation and colon cancer cell proliferation. Cancer Lett. 2019, 454, 70–77. [Google Scholar] [CrossRef]
- Zhao, G.; Song, Y.; Dong, L.; Shi, H.; Li, H.; Yang, L.; Wang, J. Silencing of lemur tyrosine kinase 2 restricts the proliferation and invasion of hepatocellular carcinoma through modulation of GSK-3β/Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 2019, 517, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Ditsiou, A.; Cilibrasi, C.; Simigdala, N.; Papakyriakou, A.; Milton-Harris, L.; Vella, V.; Nettleship, J.E.; Lo, J.H.; Soni, S.; Smbatyan, G.; et al. The structure-function relationship of oncogenic LMTK3. Sci. Adv. 2020, 6, eabc3099. [Google Scholar] [CrossRef]
- Gaozza, E.; Baker, S.J.; Vora, R.K.; Reddy, E.P. AATYK: A novel tyrosine kinase induced during growth arrest and apoptosis of myeloid cells. Oncogene 1997, 15, 3127–3135. [Google Scholar] [CrossRef]
- Wang, H.; Brautigan, D.L. A novel transmembrane Ser/Thr kinase complexes with protein phosphatase-1 and inhibitor-2. J. Biol. Chem. 2002, 277, 49605–49612. [Google Scholar] [CrossRef] [PubMed]
- Bencze, J.; Mórotz, G.M.; Seo, W.; Bencs, V.; Kálmán, J.; Miller, C.C.J.; Hortobágyi, T. Biological function of Lemur tyrosine kinase 2 (LMTK2): Implications in neurodegeneration. Mol. Brain 2018, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 9 March 2021).
- Shah, K.; Bradbury, N.A. Lemur tyrosine kinase 2, a novel target in prostate cancer therapy. Oncotarget 2015, 6, 14233–14246. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.; Jia, Y.; White, C.; Bradbury, N.A. Determination of the membrane topology of lemur tyrosine kinase 2 (LMTK2) by fluorescence protease protection. Am. J. Physiol. Cell Physiol. 2013, 304, C164–C169. [Google Scholar] [CrossRef]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef]
- Harries, L.W.; Perry, J.R.B.; McCullagh, P.; Crundwell, M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer 2010, 10, 315. [Google Scholar] [CrossRef]
- Vezelis, A.; Simiene, J.; Dabkeviciene, D.; Kincius, M.; Ulys, A.; Suziedelis, K.; Jarmalaite, S.; Jankevicius, F. LMTK2 as Potential Biomarker for Stratification between Clinically Insignificant and Clinically Significant Prostate Cancer. J. Oncol. 2021, 2021. [Google Scholar] [CrossRef]
- Vickman, R.E.; Franco, O.E.; Moline, D.C.; Vander Griend, D.J.; Thumbikat, P.; Hayward, S.W. The role of the androgen receptor in prostate development and benign prostatic hyperplasia: A review. Asian J. Urol. 2020, 7, 191–202. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of androgen receptor in prostate cancer: A review. World J. Men Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Shah, K.; Bradbury, N.A. Kinase Modulation of Androgen Receptor Signaling: Implications for Prostate Cancer. Cancer Cell Microenviron. 2015, 2, e123. [Google Scholar] [CrossRef]
- Dunn, T.A.; Chen, S.; Faith, D.A.; Hicks, J.L.; Platz, E.A.; Chen, Y.; Ewing, C.M.; Sauvageot, J.; Isaacs, W.B.; De Marzo, A.M.; et al. A novel role of myosin VI in human prostate cancer. Am. J. Pathol. 2006, 169, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Chibalina, M.V.; Arden, S.D.; Kruppa, A.J.; Kendrick-Jones, J.; Buss, F. Overexpression of myosin VI in prostate cancer cells enhances PSA and VEGF secretion, but has no effect on endocytosis. Oncogene 2010, 29, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Manser, C.; Guillot, F.; Vagnoni, A.; Davies, J.; Lau, K.F.; McLoughlin, D.M.; De Vos, K.J.; Miller, C.C.J. Lemur tyrosine kinase-2 signalling regulates kinesin-1 light chain-2 phosphorylation and binding of Smad2 cargo. Oncogene 2012, 31, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Sun, X.; Zhao, W.; Ren, M.; Zhang, C.; Wang, Z.; Xu, W. Lemur Tyrosine Kinase-3 Suppresses Growth of Prostate Cancer Via the AKT and MAPK Signaling Pathways. Cell. Physiol. Biochem. 2017, 42, 2582–2592. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Bajrami, I.; Verrill, C.; Kigozi, A.; Ouaret, D.; Aleksic, T.; Asher, R.; Han, C.; Allen, P.; Bailey, D.; et al. Dsh homolog DVL3 mediates resistance to IGFIR inhibition by regulating IGF-RAS signaling. Cancer Res. 2014, 74, 5866–5877. [Google Scholar] [CrossRef]
- Reynard, J.; Brewster, S.; Biers, S. Oxford Handbook of Urology, 3rd ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Livingstone, T.L.; Beasy, G.; Mills, R.D.; Plumb, J.; Needs, P.W.; Mithen, R.; Traka, M.H. Plant bioactives and the prevention of prostate cancer: Evidence from human studies. Nutrients 2019, 11, 2245. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Williams, M.; Sharma, H.; Chaudry, A.; Bellamy, P. A double-blind, placebo-controlled randomised trial evaluating the effect of a polyphenol-rich whole food supplement on PSA progression in men with prostate cancer—The UK NCRN Pomi-T study. Prostate Cancer Prostatic Dis. 2014, 17, 180–186. [Google Scholar] [CrossRef]
- Watson, G.; Beaver, L.; Williams, D.; Dashwood, R.; Ho, E. Phytochemicals from cruciferous vegetables, epigenetics, and prostate cancer prevention. AAPS J. 2013, 15, 951–961. [Google Scholar] [CrossRef]
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular targets of epigallocatechin—gallate (EGCG): A special focus on signal transduction and cancer. Nutrients 2018, 10, 1936. [Google Scholar] [CrossRef] [PubMed]
- Bettuzzi, S.; Brausi, M.; Rizzi, F.; Castagnetti, G.; Peracchia, G.; Corti, A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: A preliminary report from a one-year proof-of-principle study. Cancer Res. 2006, 66, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Shida, Y.; Hakariya, T.; Sakai, H. Anti-cancer effects of green tea polyphenols against prostate cancer. Molecules 2019, 24, 193. [Google Scholar] [CrossRef] [PubMed]
- Ghanavati, M.; Clark, C.C.T.; Bahrami, A.; Teymoori, F.; Movahed, M.; Sohrab, G.; Hejazi, E. Dietary intake of polyphenols and total antioxidant capacity and risk of prostate cancer: A case–control study in Iranian men. Eur. J. Cancer Care 2021, 30, e13364. [Google Scholar] [CrossRef]
- Singh, S.K.; Banerjee, S.; Acosta, E.P.; Lillard, J.W.; Singh, R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget 2017, 8, 17216–17228. [Google Scholar] [CrossRef]
- Costea, T.; Vlad, O.C.; Miclea, L.C.; Ganea, C.; Szöllősi, J.; Mocanu, M.M. Alleviation of multidrug resistance by flavonoid and non-flavonoid compounds in breast, lung, colorectal and prostate cancer. Int. J. Mol. Sci. 2020, 21, 401. [Google Scholar] [CrossRef]
- Perletti, G.; Magri, V.; Vral, A.; Stamatiou, K.; Trinchieri, A. Green tea catechins for chemoprevention of prostate cancer in patients with histologically-proven HG-PIN or ASAP. Concise review and meta-analysis. Arch. Ital. Urol. Androl. 2019, 91, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Costea, T.; Nagy, P.; Ganea, C.; Szöllősi, J.; Mocanu, M.M. Molecular mechanisms and bioavailability of polyphenols in prostate cancer. Int. J. Mol. Sci. 2019, 20, 1062. [Google Scholar] [CrossRef]
- Tauber, A.L.; Schweiker, S.S.; Levonis, S.M. From tea to treatment; epigallocatechin gallate and its potential involvement in minimizing the metabolic changes in cancer. Nutr. Res. 2020, 74, 23–36. [Google Scholar] [CrossRef]
- Shankar, S.; Suthakar, G.; Srivastava, R.K. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front. Biosci. 2007, 12, 5039–5051. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kwak, J.; Choi, H.K.; Choi, K.C.; Kim, S.; Lee, J.; Jun, W.; Park, H.J.; Yoon, H.G. EGCG suppresses prostate cancer cell growth modulating acetylation of androgen receptor by anti-histone acetyltransferase activity. Int. J. Mol. Med. 2012, 30, 69–74. [Google Scholar] [CrossRef]
- Li, M.; He, Z.; Ermakova, S.; Zheng, D.; Tang, F.; Cho, Y.Y.; Zhu, F.; Ma, W.Y.; Sham, Y.; Rogozin, E.A.; et al. Direct inhibition of insulin-like growth factor-I receptor kinase activity by (-)-epigallocatechin-3-gallate regulates cell transformation. Cancer Epidemiol. Biomark. Prev. 2007, 16, 598–605. [Google Scholar] [CrossRef]
- Shao, N.; Tang, H.; Mi, Y.; Zhu, Y.; Wan, F.; Ye, D. A novel gene signature to predict immune infiltration and outcome in patients with prostate cancer. Oncoimmunology 2020, 9, 1762473. [Google Scholar] [CrossRef] [PubMed]
- Komisarof, J.; McCall, M.; Newman, L.; Bshara, W.; Mohler, J.L.; Morrison, C.; Land, H. A four gene signature predictive of recurrent prostate cancer. Oncotarget 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Rizzi, F.; Belloni, L.; Crafa, P.; Lazzaretti, M.; Remondini, D.; Ferretti, S.; Cortellini, P.; Corti, A.; Bettuzzi, S. A novel gene signature for molecular diagnosis of human prostate cancer by RT-qPCR. PLoS ONE 2008, 3, e3617. [Google Scholar] [CrossRef]
- Ruscetti, M.A.; Wu, H. PTEN in Prostate Cancer. In Prostate Cancer Protein Reviews; Springer: New York, NY, USA, 2013; pp. 87–137. [Google Scholar]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities (Review). Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 9. [Google Scholar] [CrossRef]
- Chibalina, M.V.; Seaman, M.N.J.; Miller, C.C.; Kendrick-Jones, J.; Buss, F. Myosin VI and its interacting protein LMTK2 regulate tubule formation and transport to the endocytic recycling compartment. J. Cell Sci. 2007, 120, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Dey, I.; Bradbury, N.A. Activation of TPA-response element present in human Lemur Tyrosine Kinase 2 (lmtk2) gene increases its expression. Biochem. Biophys. Rep. 2017, 12, 140–150. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Entry Name | Protein Names | Gene Names | Length (Res.) | Proton Acceptor Active Site | ATP Binding Site | ATP Nucleotide Binding (Res. Number) |
|---|---|---|---|---|---|---|
| LMTK1_HUMAN | Serine/threonine-protein kinase LMTK1, (EC 2.7.11.1) (Apoptosis-associated tyrosine kinase) (AATYK) (Brain apoptosis-associated tyrosine kinase) (CDK5-binding protein) (Lemur tyrosine kinase 1) (p35-binding protein) (p35BP) | AATK, AATYK, KIAA0641, LMR1, LMTK1 | 1374 | D253 | K156 | 131–139 |
| LMTK2_HUMAN | Serine/threonine-protein kinase LMTK2, (EC 2.7.11.1) (Apoptosis-associated tyrosine kinase 2) (Brain-enriched kinase) (hBREK) (CDK5/p35-regulated kinase) (CPRK) (Kinase/phosphatase/inhibitor 2) (Lemur tyrosine kinase 2) (Serine/threonine-protein kinase KPI-2) | LMTK2, AATYK2, BREK, KIAA1079, KPI2, LMR2 | 1503 | D265 | K168 | 143–151 |
| LMTK3_HUMAN | Serine/threonine-protein kinase LMTK3 (EC 2.7.11.1) (Lemur tyrosine kinase 3) | LMTK3, KIAA1883, TYKLM3 | 1460 | D266 | K164 | 139–147 |
| Reference | Cell Line/Tissue | Principal Techniques | Main Results | Conclusions |
|---|---|---|---|---|
| Harries et al. [60] | Human prostate samples (cancer and benign prostatic hyperplasia, BPH) from Exeter tissue bank | PCR amplification and sequencing for genotyping 7 GWAS risk loci | Risk genotype at the GWAS variant rs6465657 correlates with LMTK2 expression | Expression levels of LMTK2 inversely correlate with the presence of prostate cancer |
| Real-time PCR | Prostate adenocarcinoma samples expressed 68% less LMTK2 mRNA than BPH samples | |||
| Vezelis et al. [61] | Blood sample of patients who had rising PSA after negative transrectal systematic prostate biopsy | Analysis of CRISP3, LMTK2 and MSMB gene expression by means of quantitative RT-PCR | LMTK2 and MSMB expression significantly decreases in blood samples of patients with PCa and Benign Prostate Disease as compared to control PSA density (ng/mL) can differentiate PCa from the benign prostate disease | PSA density, in combination with LMTK2 expression level, may assist in stratification between clinically insignificant and clinically significant PCa |
| Shah et al. [57] | Human prostate tissue array (prostate cancer, hyperplasia, and normal prostate tissue) | Immunostaining analysis | LMTK2 is down regulated in human PCa | Loss of LMTK2 protein is strongly associated with prostate cancer and prostate hyperplasia LMTK2 interacts directly with AR and inhibits its transcriptional activity LMTK2 down-regulation promotes tumour forming capacity and proliferation The decrease in LMTK2 expression in prostate cancer patient may promote tumour cells proliferation by enhancing AR transcriptional activity |
| LNCaP cells Normal human prostate tissue | Coimmunoprecipitation Colocalisation analysis by immunostaining | LMTK2 and AR interact in prostate cancer cells and colocalise in human prostate tissue | ||
| HEK293 cells | Dual luciferase assay with LMTK2 knockdown or LMTK2 overexpression | Knockdown of LMTK2 in cells expressing AR enhances androgen-dependent activation of a luciferase reporter gene Overexpression of LMTK2 in cells expressing AR decreases androgen-dependent activation of a luciferase reporter gene | ||
| LNCaP cells | Real-time PCR | LMTK2 knockdown cells, deprived from androgens, show a significant increase in mRNA expression of AR responsive genes | ||
| LNCaP cells | Tumoursphere assay Cell viability assay | LNCaP knockdown cells: -showed higher colony-forming capacity -showed ~5 times higher cell viability under androgen starvation | ||
| Puri et al. [66] | LNCaP cells | Immunofluorescence microscopy | Myosin VI is present on early endosomes, recycling endosomes and trans-Golgi network | LMTK2, together with Myosin VI, may participate in the orchestration of endosomal recycling pathway The secretory pathway via the recycling endosome can be involved in PCa pathology |
| Coimmunoprecipitation | LMTK2 binds to and coimmunoprecipitates with Myosin VI | |||
| Myosin VI siRNA knockdown | Secretion of PSA and VEGF is reduced | |||
| Manser et al. [67] | HeLa cells | Coimmunoprecipitation Immunoblot analysis | LMTK2 interacts with Protein Phosphatase-1C LMTK2 increases inhibitory phosphorylation of Glycogen Synthase Kinase-3β LMTK2 reduces Kinesin-1-Light Chain 2 phosphorylation and promotes KLC2 binding of Smad2 transcription factor | Since LMTK2 expression is significantly reduced in prostate cancer tissues, Smad2 binding to KLC2 and transport on Kinesin-1 may also be inhibited in prostate cancer cells |
| siRNA knockdown of LMTK2 | LMTK2 knockdown inhibits Smad2 nuclear signalling in response to TGF-β receptor activation |
| Reference | Cell Line/Tissue | Principal Techniques | Main Results | Conclusions |
|---|---|---|---|---|
| Sun et al. [68] | Prostate cancer tissue | Quantitative RT-PCR Immunoblot analysis data | Expression of LMTK3 is reduced as compared to normal tissue | A low level of LMTK3 expression is associated with PCa LMTK3 overexpression can induce PCa apoptosis in vitro and in vivo, and Akt and MAPK signalling pathways may contribute to this process Low levels of LMTK3 ex-pression in PCa tissue may reflect a decreased apoptotic rate |
| PC3 and LNCaP prostate cancer cells infected with recombinant lentivirus-LMTK3 | MTT and TUNEL assays Transwell and Matrigel invasion assays Immunoblot analysis | Overexpression of LMTK3:
| ||
| Subcutaneous tumour model in nude mice, based on PC3 cells infected with recombinant lentivirus-LMTK3 | Caliper measurement TUNEL assay Immunoblot analysis | Overexpression of LMTK3:
| ||
| Gao et al. [70] | DU145 prostate cancer cells | Reverse transfection with a kinase siRNA library Exposition to IGFIR1 AZ12253801 inhibitor | AZ12253801 inhibits IGFIR phosphorylation and cell viability LMTK3 depletion enhances AZ12253801 sensitivity | LMTK3 is among the putative mediators of resistance to IGFIR inhibition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, E.; Naponelli, V.; Bettuzzi, S. Lemur Tyrosine Kinases and Prostate Cancer: A Literature Review. Int. J. Mol. Sci. 2021, 22, 5453. https://doi.org/10.3390/ijms22115453
Ferrari E, Naponelli V, Bettuzzi S. Lemur Tyrosine Kinases and Prostate Cancer: A Literature Review. International Journal of Molecular Sciences. 2021; 22(11):5453. https://doi.org/10.3390/ijms22115453
Chicago/Turabian StyleFerrari, Elena, Valeria Naponelli, and Saverio Bettuzzi. 2021. "Lemur Tyrosine Kinases and Prostate Cancer: A Literature Review" International Journal of Molecular Sciences 22, no. 11: 5453. https://doi.org/10.3390/ijms22115453
APA StyleFerrari, E., Naponelli, V., & Bettuzzi, S. (2021). Lemur Tyrosine Kinases and Prostate Cancer: A Literature Review. International Journal of Molecular Sciences, 22(11), 5453. https://doi.org/10.3390/ijms22115453