
Ablation of Selenbp1 Alters Lipid Metabolism via the Pparα Pathway in Mouse Kidney

 ,
,
Abstract
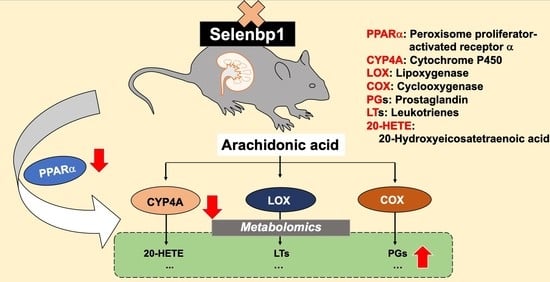
1. Introduction
2. Results
2.1. Expression Levels of Selenbp1 and Selenbp2 in the Kidney under Fasting and Non-Fasting Conditions
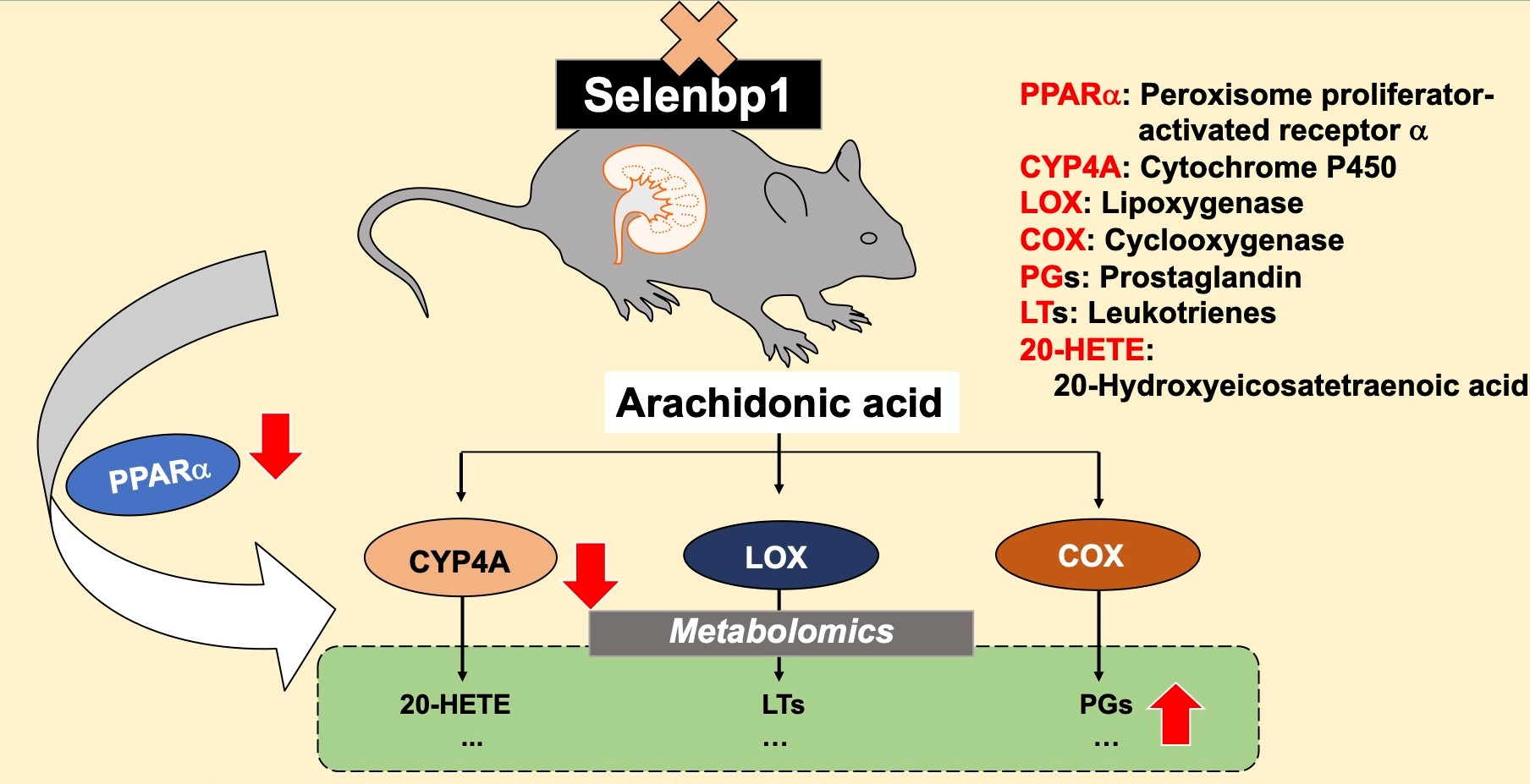
2.2. Metabolomic Changes in the Kidney of Selenbp1-KO Mice
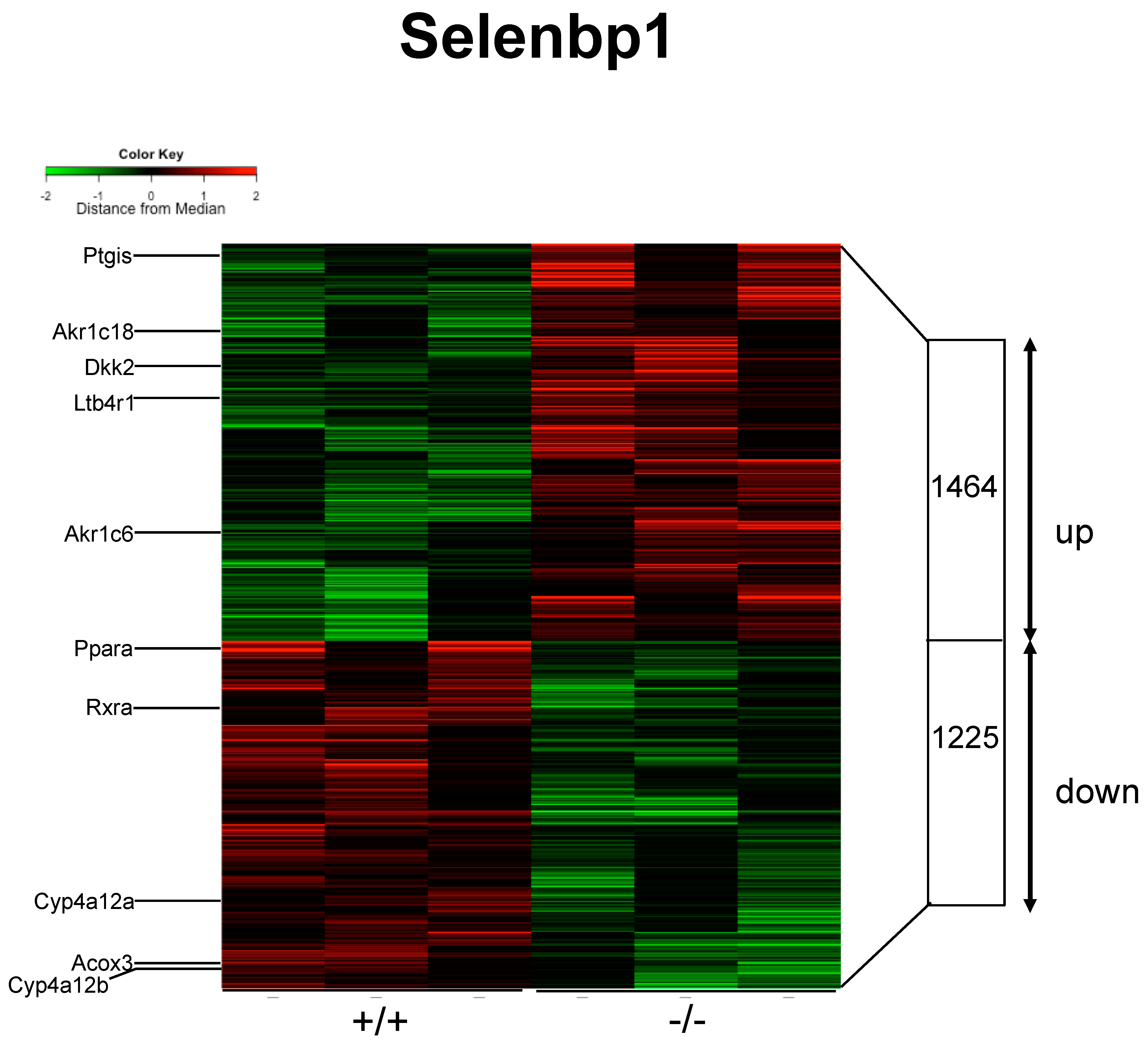
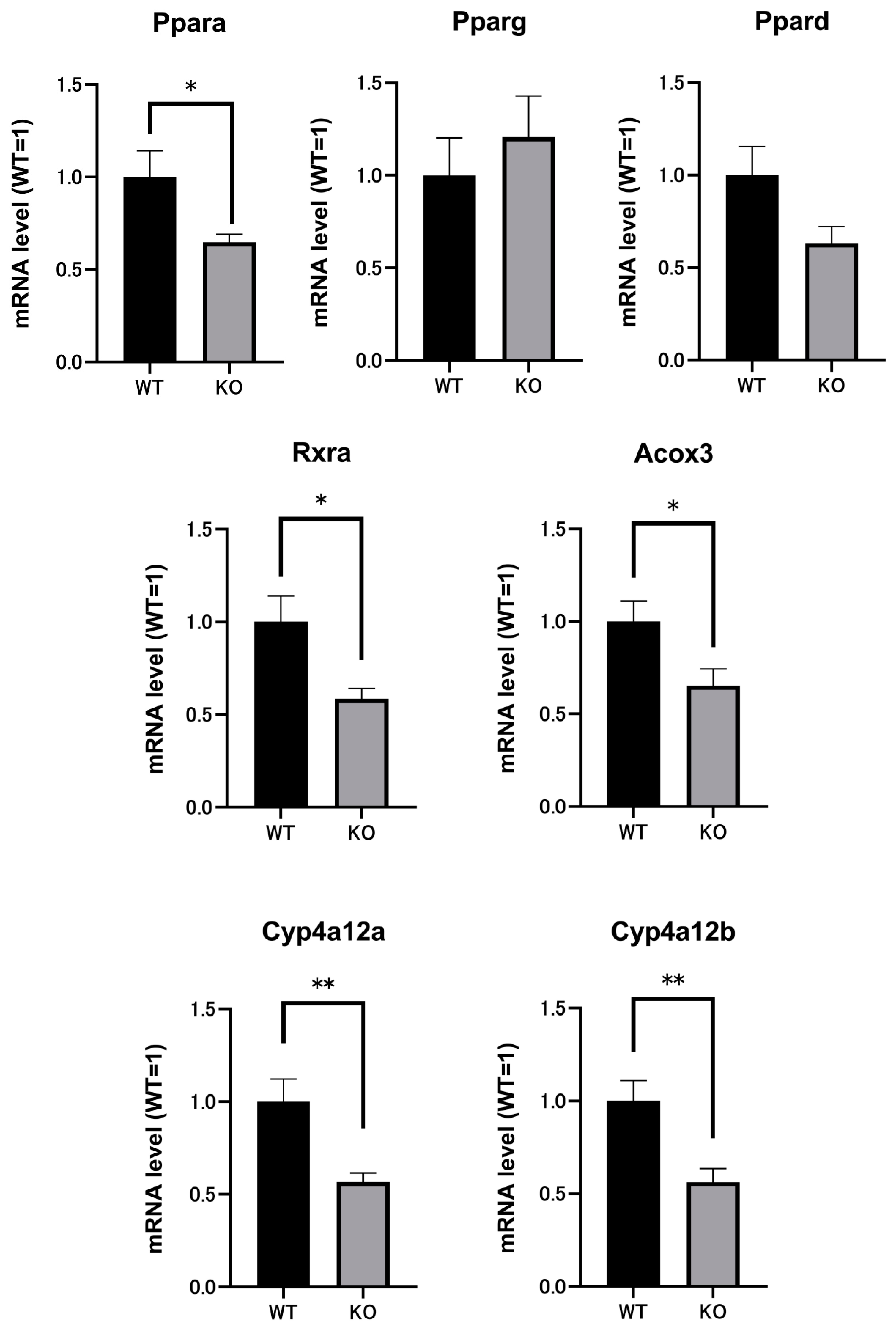
2.3. Renal mRNAs in Mice Were Altered by Selenbp1 Deletion via the Pparα Pathway
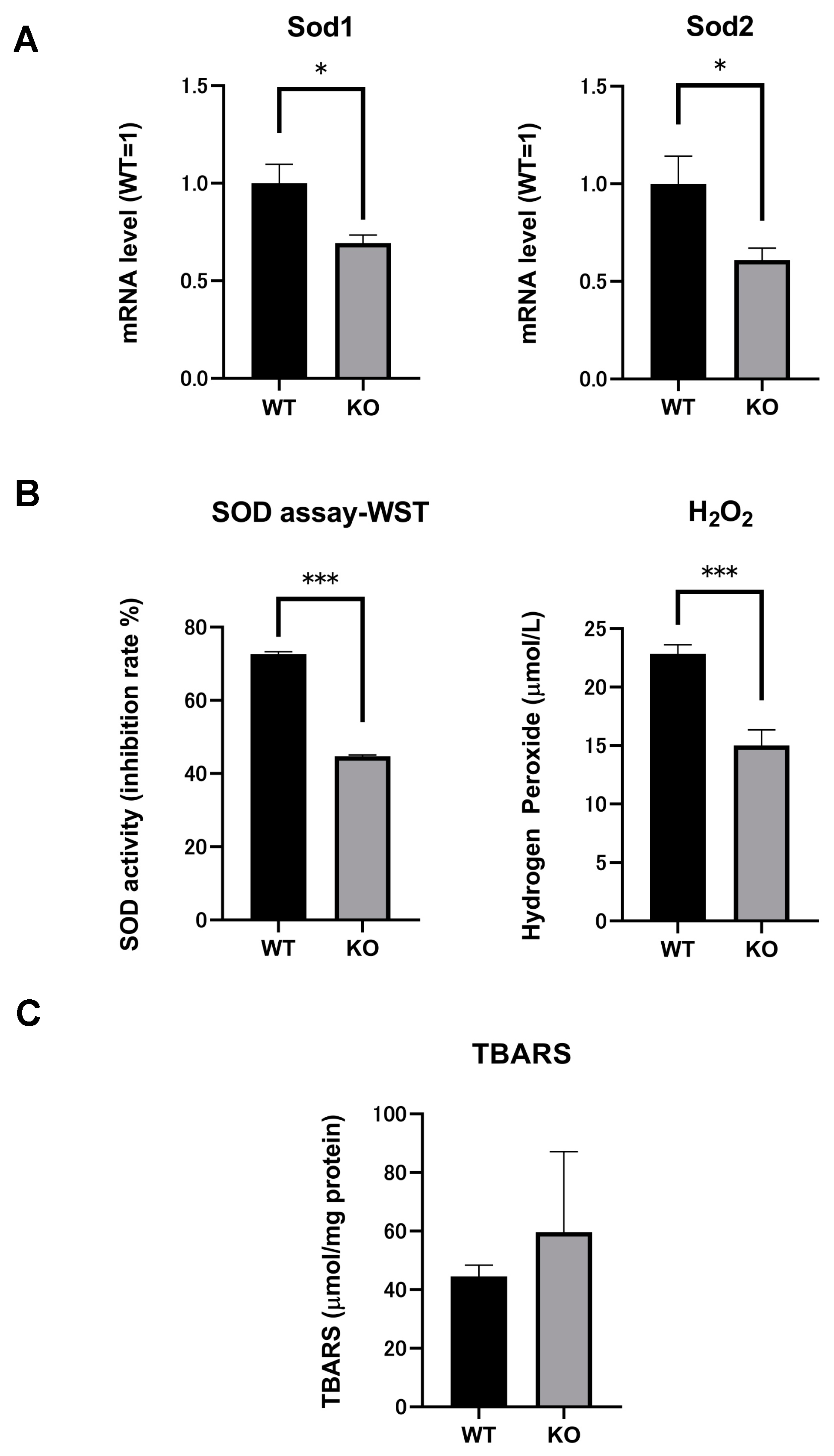
2.4. SOD mRNA and SOD Activity in the Kidneys of Both Selenbp1-KO and WT Mice
2.5. Significant Reductions in H2O2 Levels Indicated Changes in Oxidation Stress in Selenbp1-Deficient Mice
2.6. Comparison of Renal Lipid Peroxidation between Selenbp1-KO and WT Mice
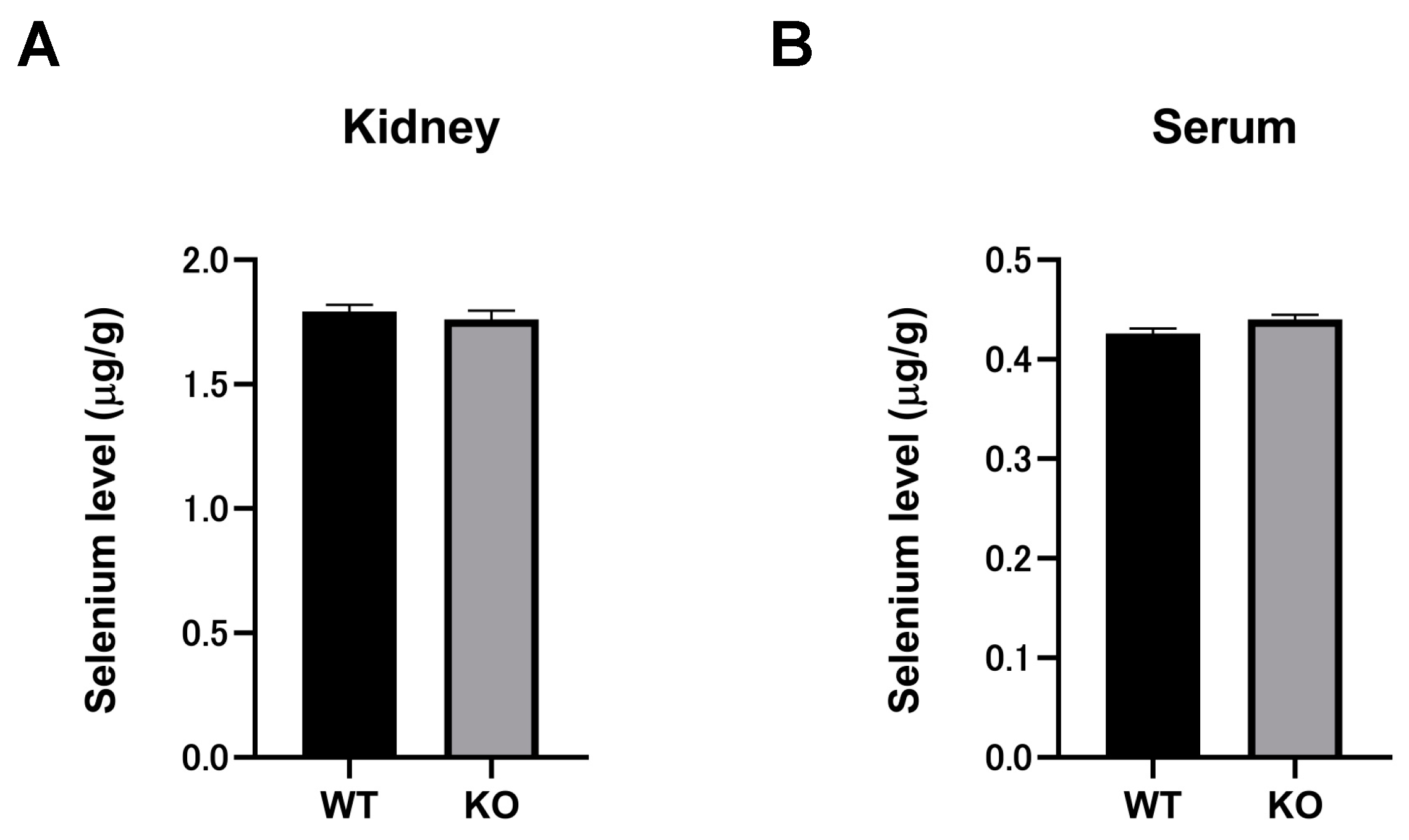
2.7. Selenium Compounds Were Unaltered in the Kidney and Serum of Selenbp1-KO Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Immunoblotting
4.3. Metabolomics of Kidneys
4.4. DNA Microarray Analysis
4.5. Quantitative Reverse Transcription (RT)-Polymerase Chain Reaction (PCR)
4.6. Superoxide Dismutase Assay with the Xanthine Oxidase/WST System
4.7. Detection of Hydrogen Peroxide
4.8. Assay for Peroxides in Mouse Kidneys Using the Thiobarbituric Acid Reaction
4.9. Quantification of Selenium Compounds by Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Arnér, E.S.; Berry, M.J.; Brigelius-Flohé, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein Gene Nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Selenium and redox signaling. Arch. Biochem. Biophys. 2017, 617, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.J. Common modifications of selenocysteine in selenoproteins. Essays Biochem. 2020, 64, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Lacourciere, G.M.; Ishii, K.; Stadtman, T.C. Characterization of potential selenium-binding proteins in the selenophosphate synthetase system. Proc. Natl. Acad. Sci. USA 2005, 102, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.P.; Oborn, C.J.; Danielson, K.G.; Medina, D. Evidence for two selenium binding proteins distinct from glutathione peroxidase in mouse liver. Carcinogenesis 1989, 10, 541–546. [Google Scholar] [CrossRef]
- Bansal, M.P.; Mukhopadhyay, T.; Scott, J.; Cook, R.G.; Mukhopadhyay, R.; Medina, D. DNA sequencing of a mouse liver protein that binds selenium- implications for selenium’s mechanism of action in cancer prevention. Carcinogenesis 1990, 11, 2071–2073. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Micoogullari, M.; Hoang, N.A.; Bergheim, I.; Klotz, L.-O.; Sies, H. Selenium-binding protein 1 (SELENBP1) is a marker of mature adipocytes. Redox Biol. 2019, 20, 489–495. [Google Scholar] [CrossRef]
- Pol, A.; Renkema, G.H.; Tangerman, A.; Winkel, E.G.; Engelke, U.F.; de Brouwer, A.P.M.; Lloyd, K.C.; Araiza, R.S.; van den Heuvel, L.; Omran, H.; et al. Mutations in SELENBP1, encoding a novel human methanethiol oxidase, cause extraoral halitosis. Nat. Genet. 2018, 50, 120–129. [Google Scholar] [CrossRef]
- Chen, G.; Wang, H.; Miller, C.T.; Thomas, D.G.; Gharib, T.G.; Misek, D.E.; Giordano, T.J.; Orringer, M.B.; Hanash, S.M.; Beer, D.G. Reduced selenium-binding protein 1 expression is associated with poor outcome in lung adenocarcinomas. J. Pathol. 2004, 202, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, W.; Li, M.; Byun, D.S.; Tong, C.; Nasser, S.; Zhuang, M.; Arango, D.; Mariadason, J.M.; Augenlicht, L.H. Expression of selenium-binding protein 1 characterizes intestinal cell maturation and predicts survival for patients with colorectal cancer. Mol. Nutr. Food Res. 2008, 52, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Schott, M.; de Jel, M.M.; Engelmann, J.C.; Renner, P.; Geissler, E.K.; Bosserhoff, A.K.; Kuphal, S. Selenium-binding protein 1 is down-regulated in malignant melanoma. Oncotarget 2018, 9, 10445–10456. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.J.; Everall, I.P.; Kremen, W.S.; Corbeil, J.; Sásik, R.; Khanlou, N.; Han, M.; Liew, C.C.; Tsuang, M.T. Comparative gene expression analysis of blood and brain provides concurrent validation of selenbp1 up-regulation in schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 15533–15538. [Google Scholar] [CrossRef] [PubMed]
- Porat, A.; Sagiv, Y.; Elazar, Z. A 56-kDa selenium-binding protein participates in intra-Golgi protein transport. J. Biol. Chem. 2000, 275, 14457–14465. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Wang, Y.; Sytkowski, A.J. Human selenium binding protein-1 (hSP56) interacts with VDU1 in a selenium-dependent manner. Biochem. Biophys. Res. Commun. 2009, 379, 583–588. [Google Scholar] [CrossRef]
- Miyaguchi, K. Localization of Selenium-binding protein at the tips of rapidly extending protrusions. Histochem. Cell. Biol. 2004, 121, 371–376. [Google Scholar] [CrossRef]
- Jamba, L.; Nehru, B.; Bansal, M.P. Redox modulation of selenium binding proteins by cadmium exposures in mice. Mol. Cell Biochem. 1997, 177, 169–175. [Google Scholar] [CrossRef]
- Lanfear, J.; Fleming, J.; Walker, M.; Harrison, P. Different patterns of regulation of the genes encoding the closely related 56 kDa selenium-and acetaminophen-binding proteins in normal tissues and during carcinogenesis. Carcinogenesis 1993, 14, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Pumford, N.R.; Martin, B.M.; Hinson, J.A. A metabolite of acetaminophen covalently binds to the 56 KDa selenium binding protein. Biochem. Biophys. Res. Commun. 1992, 182, 1348–1355. [Google Scholar] [CrossRef]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Reyes, H.; Reisz-Porszasz, S.; Hankinson, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992, 256, 1193–1195. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.M.; Hilbert, D.M.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996, 140, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Hatsumura, M.; Ishida, T.; Ariyoshi, N.; Oguri, K. Significant induction of a 54-kDa protein in rat liver with homologous alignment to mouse selenium binding protein by a coplanar polychlorinated biphenyl, 3,4,5,3′,4′-pentachlorobiphenyl and 3-methylcholanthrene. Toxicol. Lett. 1996, 87, 1–9. [Google Scholar] [CrossRef]
- Tsujimoto, S.; Ishida, T.; Takeda, T.; Ishii, Y.; Onomura, Y.; Tsukimori, K.; Takechi, S.; Yamaguchi, T.; Uchi, H.; Suzuki, S.O.; et al. Selenium-binding protein 1: Its physiological function, dependence on aryl hydrocarbon receptors, and role in wasting syndrome by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochim. Biophys. Acta. 2013, 1830, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.C.; Gijón, M.A. Biosynthesis and metabolism of leukotrienes. Biochem. J. 2007, 405, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, G.; Erb, W.; Aggarwal, V.K. Stereocontrolled organocatalytic synthesis of prostaglandin PGF2α in seven steps. Nature 2012, 489, 278–281. [Google Scholar] [CrossRef]
- Figueiredo-Pereira, M.E.; Corwin, C.; Babich, J. Prostaglandin J2: A potential target for halting inflammation-induced neurodegeneration. Ann. N. Y. Acad. Sci. 2016, 1363, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, H.; Zhao, J.; Dai, Q.; Huang, C.; Jin, L.; Yang, F.; Chen, F.; Wang, O.; Gao, Y. Plasma ω-3 and ω-6 fatty acids in thyroid diseases. Oncol. Lett. 2018, 16, 5433–5440. [Google Scholar] [CrossRef]
- Asztalos, I.B.; Gleason, J.A.; Sever, S.; Gedik, R.; Asztalos, B.F.; Horvath, K.V.; Dansinger, M.L.; Lamon-Fava, S.; Schaefer, E.J. Effects of eicosapentaenoic acid and docosahexaenoic acid on cardiovascular disease risk factors: A randomized clinical trial. Metabolism 2016, 65, 1636–1645. [Google Scholar] [CrossRef]
- Capdevila, J.; Chacos, N.; Werringloer, J.; Prough, R.A.; Estabrook, R.W. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc. Natl. Acad. Sci. USA 1981, 78, 5362–5366. [Google Scholar] [CrossRef]
- Johnson, A.L.; Edson, K.Z.; Totah, R.A.; Rettie, A.E. Cytochrome P450 ω-Hydroxylases in Inflammation and Cancer. Adv. Pharmacol. 2015, 74, 223–262. [Google Scholar] [CrossRef] [PubMed]
- Bardot, O.; Aldridge, T.C.; Latruffe, N.; Green, S. PPAR-RXR heterodimer activates a peroxisome proliferator response element upstream of the bifunctional enzyme gene. Biochem. Biophys. Res. Commun. 1993, 192, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, A.; Kong, L.; Zhang, Q.; Ling, E. Functional analysis of insect molting fluid proteins on the protection and regulation of ecdysis. J. Biol. Chem. 2014, 289, 35891–35906. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Yang, Q. Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J. Cardiol. 2013, 5, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kunau, W.H.; Dommes, V.; Schulz, H. Beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: A century of continued progress. Prog. Lipid Res. 1995, 34, 267–342. [Google Scholar] [CrossRef]
- Kamijo, Y.; Hora, K.; Kono, K.; Takahashi, K.; Higuchi, M.; Ehara, T.; Kiyosawa, K.; Shigematsu, H.; Gonzalez, F.J.; Aoyama, T. PPARalpha protects proximal tubular cells from acute fatty acid toxicity. J. Am. Soc. Nephrol. 2007, 18, 3089–3100. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Lee, E.K.; Shin, Y.J.; Park, E.Y.; Kim, N.D.; Moon, A.; Kwack, S.J.; Son, J.Y.; Kacew, S.; Lee, B.M.; Bae, O.N.; et al. Selenium-binding protein 1: A sensitive urinary biomarker to detect heavy metal-induced nephrotoxicity. Arch. Toxicol. 2017, 91, 1635–1648. [Google Scholar] [CrossRef]
- Hatsumura, M.; Ishida, T.; Ishii, Y.; Ariyoshi, N.; Oguri, K.; Yoshimura, H. Effect of a coplanar PCB on lipid metabolism: The remarkable difference between rats and guinea pigs. Fukuoka Igaku Zasshi 1995, 86, 135–143. [Google Scholar] [PubMed]
- Rae, S.A.; Davidson, E.M.; Smith, M.J. Leukotriene B4, an inflammatory mediator in gout. Lancet 1982, 2, 1122–1124. [Google Scholar] [CrossRef]
- Rand, A.A.; Barnych, B.; Morisseau, C.; Cajka, T.; Lee, K.S.S.; Panigrahy, D.; Hammock, B.D. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc. Natl. Acad. Sci. USA 2017, 114, 4370–4375. [Google Scholar] [CrossRef]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front Mol. Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef]
- Liston, T.E.; Roberts, L.J., 2nd. Transformation of prostaglandin D2 to 9 alpha, 11 beta-(15S)-trihydroxyprosta-(5Z,13E)-dien-1-oic acid (9 alpha, 11 beta-prostaglandin F2): A unique biologically active prostaglandin produced enzymatically in vivo in humans. Proc. Natl. Acad. Sci. USA 1985, 82, 6030–6034. [Google Scholar] [CrossRef] [PubMed]
- Koda, N.; Tsutsui, Y.; Niwa, H.; Ito, S.; Woodward, D.F.; Watanabe, K. Synthesis of prostaglandin F ethanolamide by prostaglandin F synthase and identification of Bimatoprost as a potent inhibitor of the enzyme: New enzyme assay method using LC/ESI/MS. Arch. Biochem. Biophys. 2004, 424, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Pastel, E.; Pointud, J.C.; Loubeau, G.; Dani, C.; Slim, K.; Martin, G.; Volat, F.; Sahut-Barnola, I.; Val, P.; Martinez, A.; et al. Aldose reductases influence prostaglandin F2α levels and adipocyte differentiation in male mouse and human species. Endocrinology 2015, 156, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.A.; Kiprowska, M.; Jean-Louis, T.; Rockwell, P.; Figueiredo-Pereira, M.E.; Serrano, P.A. PACAP27 mitigates an age-dependent hippocampal vulnerability to PGJ2-induced spatial learning deficits and neuroinflammation in mice. Brain Behav. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Mayatepek, E.; Hoffmann, G.F. Leukotrienes: Biosynthesis, metabolism, and pathophysiologic significance. Pediatr. Res. 1995, 37, 1–9. [Google Scholar] [CrossRef]
- Kikuta, Y.; Kato, M.; Yamashita, Y.; Miyauchi, Y.; Tanaka, K.; Kamada, N.; Kusunose, M. Human leukotriene B4 omega-hydroxylase (CYP4F3) gene: Molecular cloning and chromosomal localization. DNA Cell. Biol. 1998, 17, 221–230. [Google Scholar] [CrossRef]
- Sutyak, J.; Austen, K.F.; Soberman, R.J. Identification of an aldehyde dehydrogenase in the microsomes of human polymorphonuclear leukocytes that metabolizes 20-aldehyde leukotriene B4. J. Biol. Chem. 1989, 264, 14818–14823. [Google Scholar] [CrossRef]
- Baumert, T.; Huber, M.; Mayer, D.; Keppler, D. Ethanol-induced inhibition of leukotriene degradation by omega-oxidation. Eur. J. Biochem. 1989, 182, 223–229. [Google Scholar] [CrossRef]
- Lasker, J.M.; Chen, W.B.; Wolf, I.; Bloswick, B.P.; Wilson, P.D.; Powell, P.K. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. J. Biol. Chem. 2000, 275, 4118–4126. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.J.; Bammler, T.; Wolf, C.R. Deduced amino acid sequence of a murine cytochrome P-450 Cyp4a protein: Developmental and hormonal regulation in liver and kidney. Biochim. Biophys. Acta 1994, 1200, 182–190. [Google Scholar] [CrossRef]
- Wu, C.C.; Mei, S.; Cheng, J.; Ding, Y.; Weidenhammer, A.; Garcia, V.; Zhang, F.; Gotlinger, K.; Manthati, V.L.; Falck, J.R.; et al. Androgen-sensitive hypertension associates with upregulated vascular CYP4A12-20-HETE synthase. J. Am. Soc. Nephrol. 2013, 24, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fu, X.; Chen, Q.; Patra, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic Acid Metabolism and Kidney Inflammation. Int. J. Mol. Sci. 2019, 20, 3683. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Guan, Y.; Breyer, M.D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2001, 60, 14–30. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Yook, P.; Costet, P.; Bianchi, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J. Biol. Chem. 1998, 273, 31581–31589. [Google Scholar] [CrossRef] [PubMed]
- Leid, M.; Kastner, P.; Chambon, P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem. Sci. 1992, 17, 427–433. [Google Scholar] [CrossRef]
- Westin, M.A.; Hunt, M.C.; Alexson, S.E. Peroxisomes contain a specific phytanoyl-CoA/pristanoyl-CoA thioesterase acting as a novel auxiliary enzyme in alpha- and beta-oxidation of methyl-branched fatty acids in mouse. J. Biol. Chem. 2007, 282, 26707–26716. [Google Scholar] [CrossRef] [PubMed]
- Ueta, N. Biochemistry of Branched Chain Fatty Acids. J-Stage 1971, 20, 663–669. [Google Scholar] [CrossRef][Green Version]
- Zhao, C.; Zeng, H.; Wu, R.T.; Cheng, W.H. Loss of Selenium-Binding Protein 1 Decreases Sensitivity to Clastogens and Intracellular Selenium Content in HeLa Cells. PLoS ONE 2016, 11, e0158650. [Google Scholar] [CrossRef]
- Liu, X.; Jang, S.S.; An, Z.; Song, H.; Kim, W.D.; Yu, J.R.; Park, W.Y. Fenofibrate decreases radiation sensitivity via peroxisome proliferator-activated receptor α-mediated superoxide dismutase induction in HeLa cells. Radiat. Oncol. J. 2012, 30, 88–95. [Google Scholar] [CrossRef]
- McAdam, E.; Brem, R.; Karran, P. Oxidative Stress-Induced Protein Damage Inhibits DNA Repair and Determines Mutation Risk and Therapeutic Efficacy. Mol. Cancer Res. 2016, 14, 612–622. [Google Scholar] [CrossRef]
- Takeda, T.; Komiya, Y.; Koga, T.; Ishida, T.; Ishii, Y.; Kikuta, Y.; Nakaya, M.; Kurose, H.; Yokomizo, T.; Shimizu, T.; et al. Dioxin-induced increase in leukotriene B4 biosynthesis through the aryl hydrocarbon receptor and its relevance to hepatotoxicity owing to neutrophil infiltration. J. Biol. Chem. 2017, 292, 10586–10599. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, P.; Zarrilli, V.; Varricchio, E.; Tramontano, D.; Mancini, F.P. The dietary antioxidant resveratrol affects redox changes of PPARalpha activity. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 247–256. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Jandrić, Z.; Roberts, D.; Rathor, M.N.; Abrahim, A.; Islam, M.; Cannavan, A. Assessment of fruit juice authenticity using UPLC-QToF MS: A metabolomics approach. Food Chem. 2014, 148, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Holmes, E.; Lundstedt, T. Chemometrics in metabonomics. J. Proteome Res. 2007, 6, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.; Carey, V.J.; Huber, W.; Irizarry, R.A.; Dudoit, S. Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Matsumoto, Y.; Ishida, T.; Takeda, T.; Koga, T.; Fujii, M.; Ishii, Y.; Fujimura, Y.; Miura, D.; Wariishi, H.; Yamada, H. Maternal exposure to dioxin reduces hypothalamic but not pituitary metabolome in fetal rats: A possible mechanism for a fetus-specific reduction in steroidogenesis. J. Toxicol. Sci. 2010, 35, 365–373. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Metabolic Pathways | Retention Time (min) | m/z | Metabolite | X-Fold | p-Value |
|---|---|---|---|---|---|---|
| Selenbp1-KO/Wild-type | Steroid metabolism | 1.56 1.42 | 347.1458 345.1872 | 19-Noraldosterone 11-Dehydrocorticosterone | 3.6 12.4 | 0.0115 0.0073 |
| Prostaglandin and leukotriene metabolism | 6.89 1.04 | 335.1496 355.1588 | Prostaglandin J2 11-Epi-prostaglandin F2α | 3.7 4.6 | 0.0161 0.0056 | |
| Fatty acid biosynthesis | 1.43 1.05 | 331.2071 303.0772 | Docosapentaenoic acid 3-Hydroxycapric acid | 1.7 1.8 | 0.0139 0.0335 | |
| Fatty acid ester biosynthesis | 1.71 | 435.3324 | Butyl oleate sulfate | 24.0 | 0.0498 | |
| Ubiquinone biosynthesis | 2.01 | 451.3326 | Vitamin K1 | 5.0 | 0.0156 | |
| Wild-type /Selenbp1 -KO | Sex hormone metabolism | 1.33 | 289.1626 | 4-Hydroxyestradiol | 4.0 | 0.0269 |
| Prostaglandin and leukotriene metabolism | 1.41 1.63 | 367.1941 353.181 | 20-Carboxy-leukotriene B4 20-Dihydroxy-leukotriene B4 | 16.9 4.4 | 0.0128 0.0165 | |
| Steroid lipid | 1.07 | 585.3781 | Cholic acid glucuronide | 4.0 | 0.0042 |
| Name | Gene ID | Comparison Ratio | p-Value | |
|---|---|---|---|---|
| 1 | Rxrα (retinoid X receptor alpha) | 20181 | 0.873 | 0.0261 |
| 2 | Acox3 (acyl-coenzyme A oxidase 3) | 80911 | 0.688 | 0.0017 |
| 3 | Cyp4a12a (cytochrome P450, family 4) | 277753 | 0.684 | 0.0042 |
| 4 | Cyp4a12b (cytochrome P450, family 4) | 13118 | 0.672 | 0.0029 |
| 5 | Pparα (peroxisome proliferator-activated receptor alpha) | 19013 | 0.792 | 0.0297 |
| 6 | Cpt1a (carnitine palmitoyl transferase 1a) | 12894 | 0.834 | 0.0194 |
| 7 | Cyp2e1 (cytochrome P450, family 2, subfamily e, polypeptide 1) | 13106 | 0.883 | 0.0488 |
| 8 | Cyp2a4 (cytochrome P450, family 2, subfamily a, polypeptide 4) | 13086 | 0.741 | 0.0110 |
| 9 | Cyp2a5 (cytochrome P450, family 2, subfamily a, polypeptide 5) | 13087 | 0.736 | 0.0078 |
| 10 | Slc51a (solute carrier family 51, alpha subunit) | 106407 | 0.864 | 0.0290 |
| 11 | Fads2 (fatty acid desaturase 2) | 56473 | 0.518 | 0.0037 |
| 12 | Dkk2 (dickkopf homolog 2) | 56811 | 1.337 | 0.0048 |
| 13 | Bpifa1 (BPI fold containing family A member 1) | 18843 | 1.190 | 0.0453 |
| 14 | Fabp6 (fatty acid-binding protein 6) | 16204 | 2.236 | 0.0420 |
| 15 | Lpcat2 (lysophosphatidylcholine acyltransferase 2) | 270084 | 1.240 | 0.0423 |
| 16 | Alox5 (arachidonate 5-lipoxygenase) | 11689 | 1.949 | 0.0194 |
| 17 | Ltb4r1 (leukotriene B4 receptor1) | 16995 | 1.372 | 0.0287 |
| 18 | Ptgis (prostaglandin I2 synthase) | 19223 | 1.359 | 0.0374 |
| 19 | Ptges3l (prostaglandin E synthase 3-like) | 56351 | 1.267 | 0.0209 |
| 20 | Akr1c6 (aldo-keto reductase family 1 member C6) | 83702 | 1.746 | 0.0442 |
| 21 | Akr1c14 (aldo-keto-reductase family 1 member C14) | 105387 | 1.144 | 0.0460 |
| 22 | Akr1c18 (aldo-keto-reductase family 1 member C18) | 105349 | 1.520 | 0.0057 |
| 23 | Akr1c 20 (aldo-keto-reductase family 1 member C20) | 116852 | 1.367 | 0.0115 |
| 24 | Slc25a23 (solute carrier family 25, member 23) | 66972 | 1.221 | 0.0184 |
| 25 | Slc25a19 (solute carrier family 25, member 19) | 67283 | 1.182 | 0.0316 |
| 26 | Slc2a3 (solute carrier family, member 3) | 20527 | 1.723 | 0.0167 |
| 27 | Slc11a1(solute carrier family 11, member 1) | 18173 | 1.295 | 0.0058 |
| 28 | Pltp (phospholipid transfer protein) | 18830 | 1.534 | 0.0452 |
| 29 | Acsbg1 (acyl-CoA synthetase bubblegum family member 1) | 94180 | 1.302 | 0.0073 |
| 30 | Angptl6 (angiopoietin-like 6) | 70726 | 1.221 | 0.0157 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Kurose, A.; Li, R.; Takeda, T.; Onomura, Y.; Koga, T.; Mutoh, J.; Ishida, T.; Tanaka, Y.; Ishii, Y. Ablation of Selenbp1 Alters Lipid Metabolism via the Pparα Pathway in Mouse Kidney. Int. J. Mol. Sci. 2021, 22, 5334. https://doi.org/10.3390/ijms22105334
Song Y, Kurose A, Li R, Takeda T, Onomura Y, Koga T, Mutoh J, Ishida T, Tanaka Y, Ishii Y. Ablation of Selenbp1 Alters Lipid Metabolism via the Pparα Pathway in Mouse Kidney. International Journal of Molecular Sciences. 2021; 22(10):5334. https://doi.org/10.3390/ijms22105334
Chicago/Turabian StyleSong, Yingxia, Atsushi Kurose, Renshi Li, Tomoki Takeda, Yuko Onomura, Takayuki Koga, Junpei Mutoh, Takumi Ishida, Yoshitaka Tanaka, and Yuji Ishii. 2021. "Ablation of Selenbp1 Alters Lipid Metabolism via the Pparα Pathway in Mouse Kidney" International Journal of Molecular Sciences 22, no. 10: 5334. https://doi.org/10.3390/ijms22105334
APA StyleSong, Y., Kurose, A., Li, R., Takeda, T., Onomura, Y., Koga, T., Mutoh, J., Ishida, T., Tanaka, Y., & Ishii, Y. (2021). Ablation of Selenbp1 Alters Lipid Metabolism via the Pparα Pathway in Mouse Kidney. International Journal of Molecular Sciences, 22(10), 5334. https://doi.org/10.3390/ijms22105334