
Transient Excursions to Membrane Core as Determinants of Influenza Virus Fusion Peptide Activity


Abstract
1. Introduction
2. Materials and Methods
2.1. Computational Methods
2.1.1. Simulated Systems
2.1.2. MD Simulations
2.1.3. Kinetic Analysis
2.1.4. Free Energy Calculations
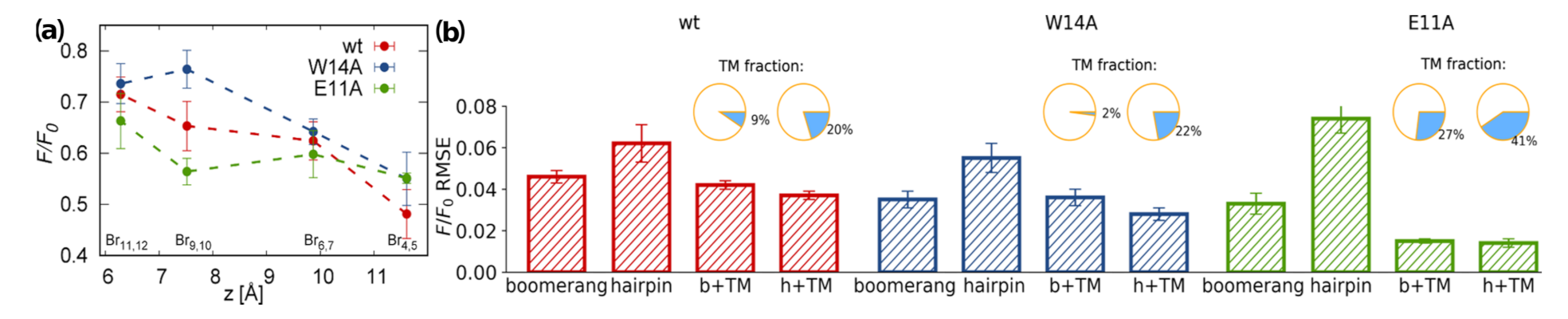
2.1.5. Tryptophan Fluorescence Calculations
2.1.6. Membrane Perturbation
2.1.7. Peptide Supervised Insertion
2.2. Experimental Methods
2.2.1. Materials
2.2.2. Liposome Preparation
2.2.3. Lipid Mixing
2.2.4. Tryptophan Fluorescence
2.2.5. Tryptophan Quenching
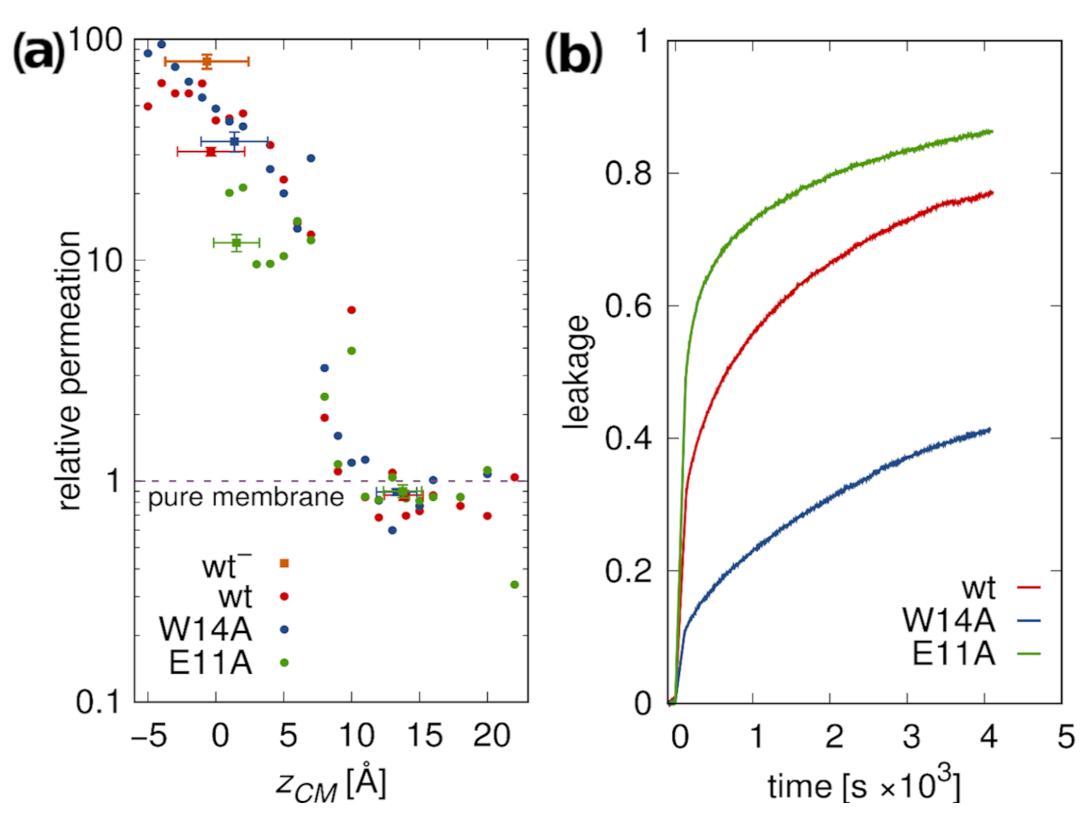
2.3. Leakage Assay
3. Results
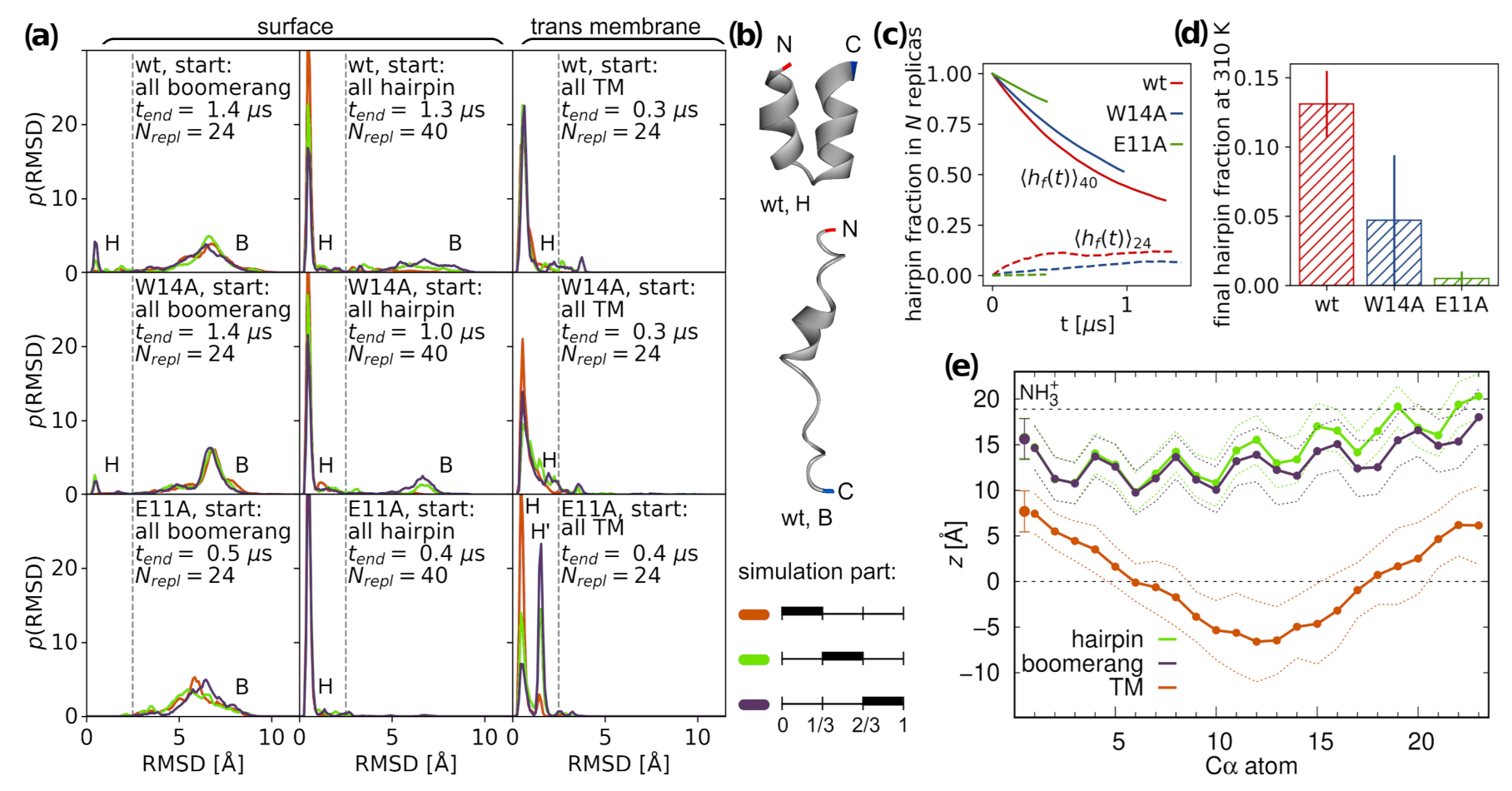
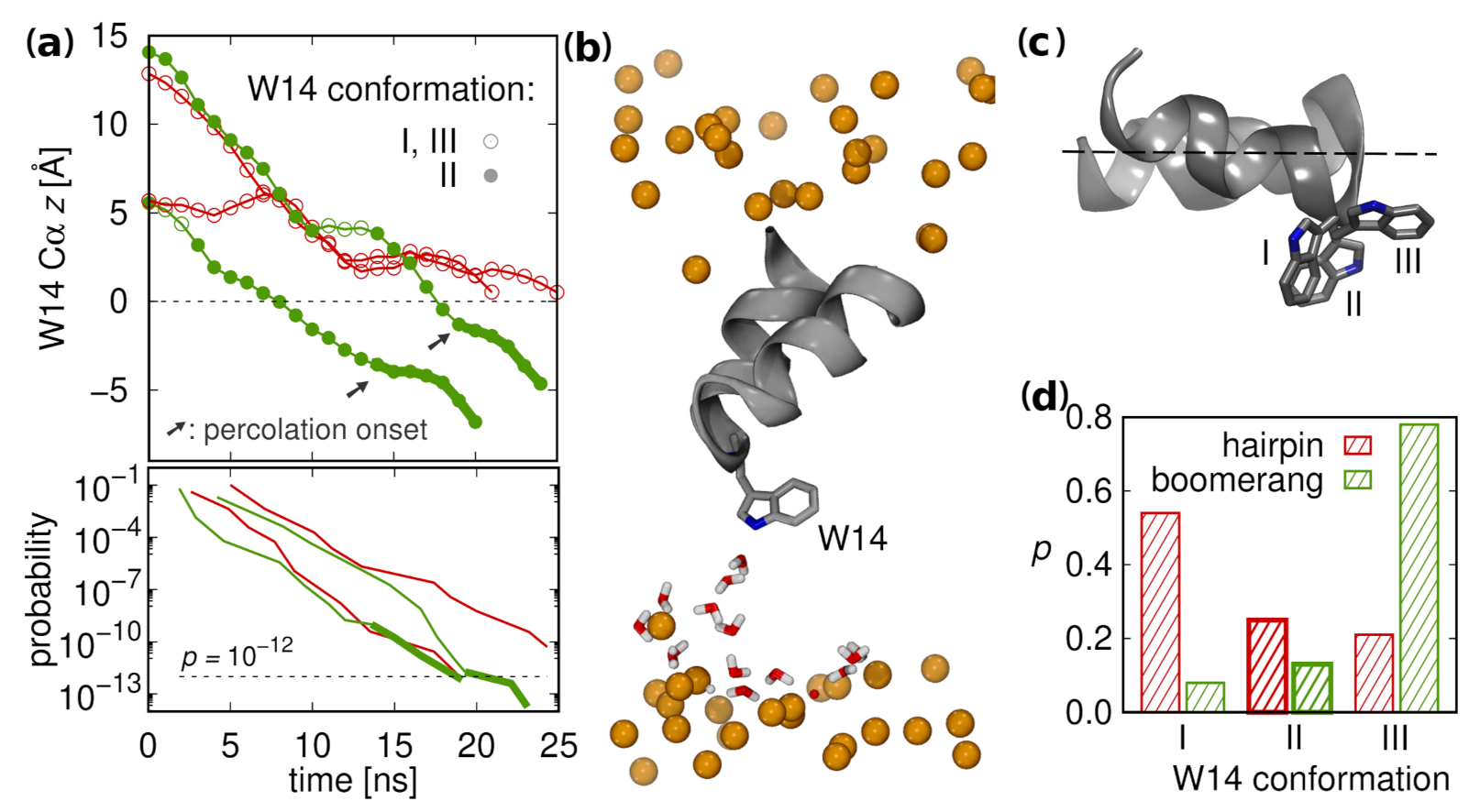
3.1. Peptide Configurations
3.1.1. Surface Placement
3.1.2. Intramembrane Placement
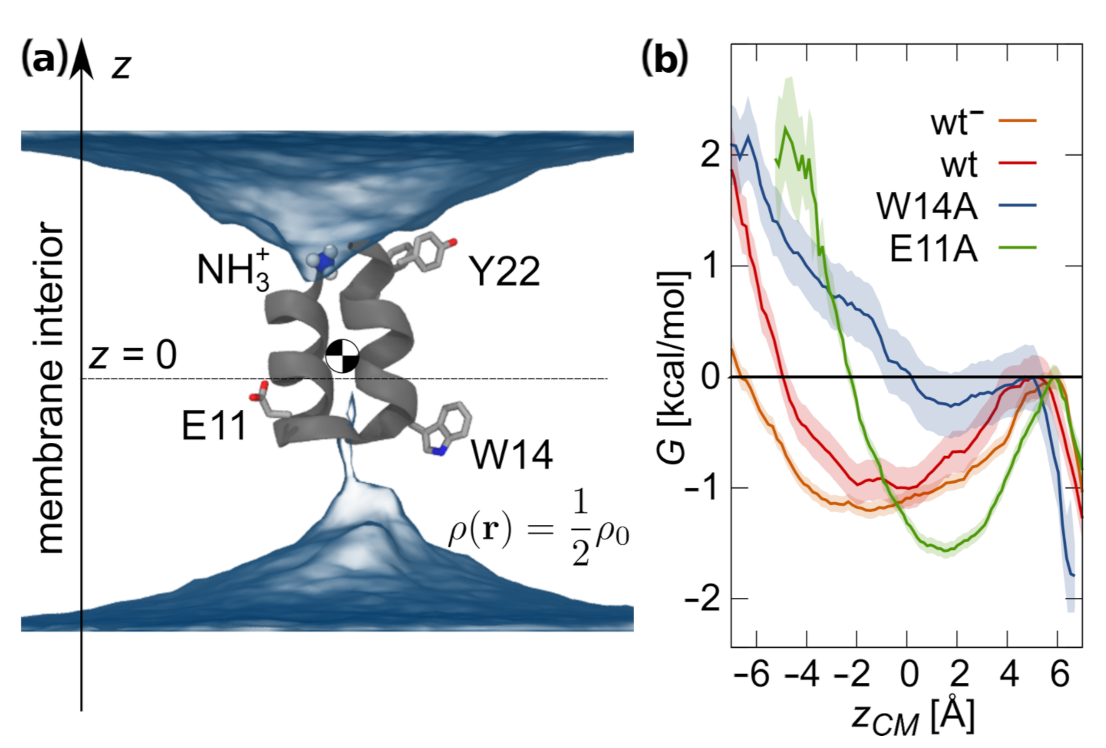
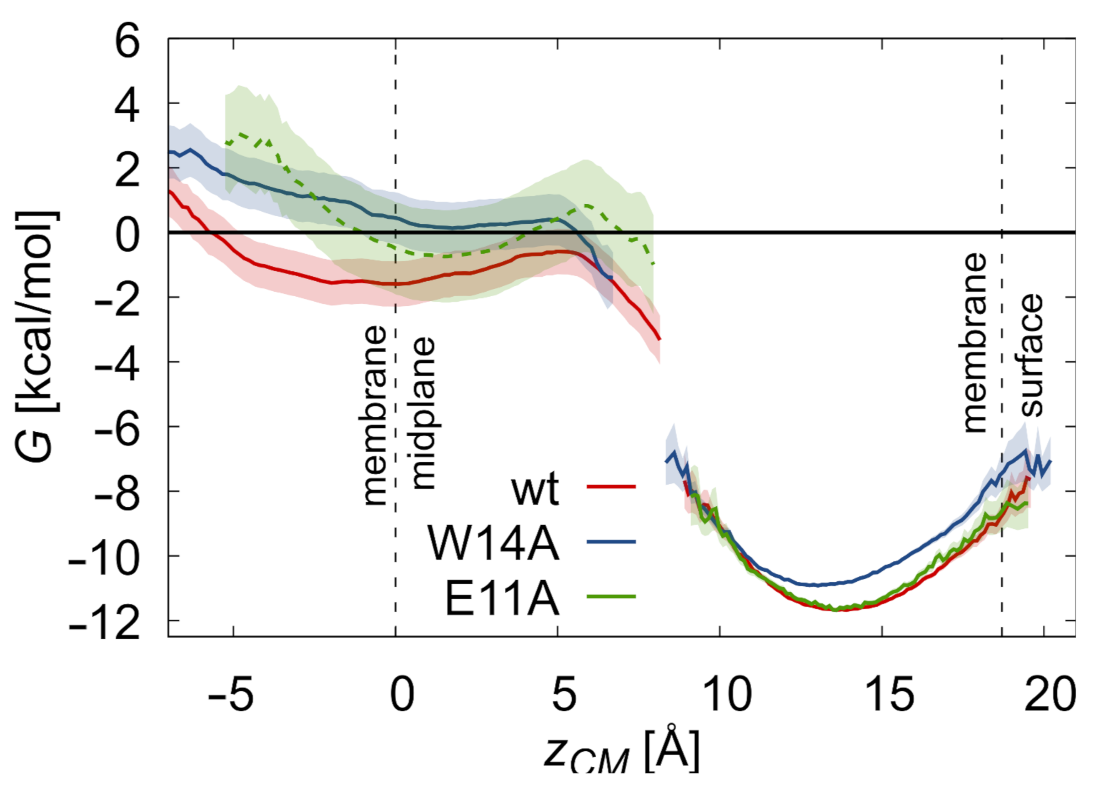
3.2. Potentials of Mean Force
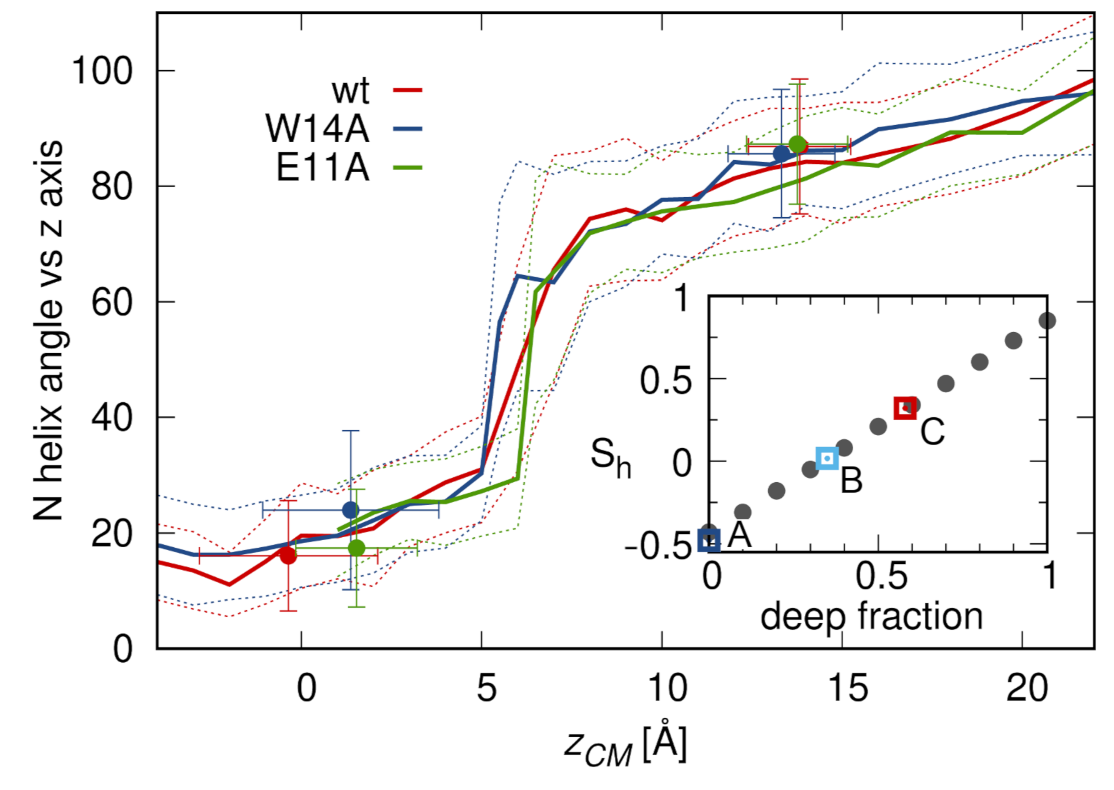
3.3. N-Helix Orientation
3.4. TRP Fluorescence
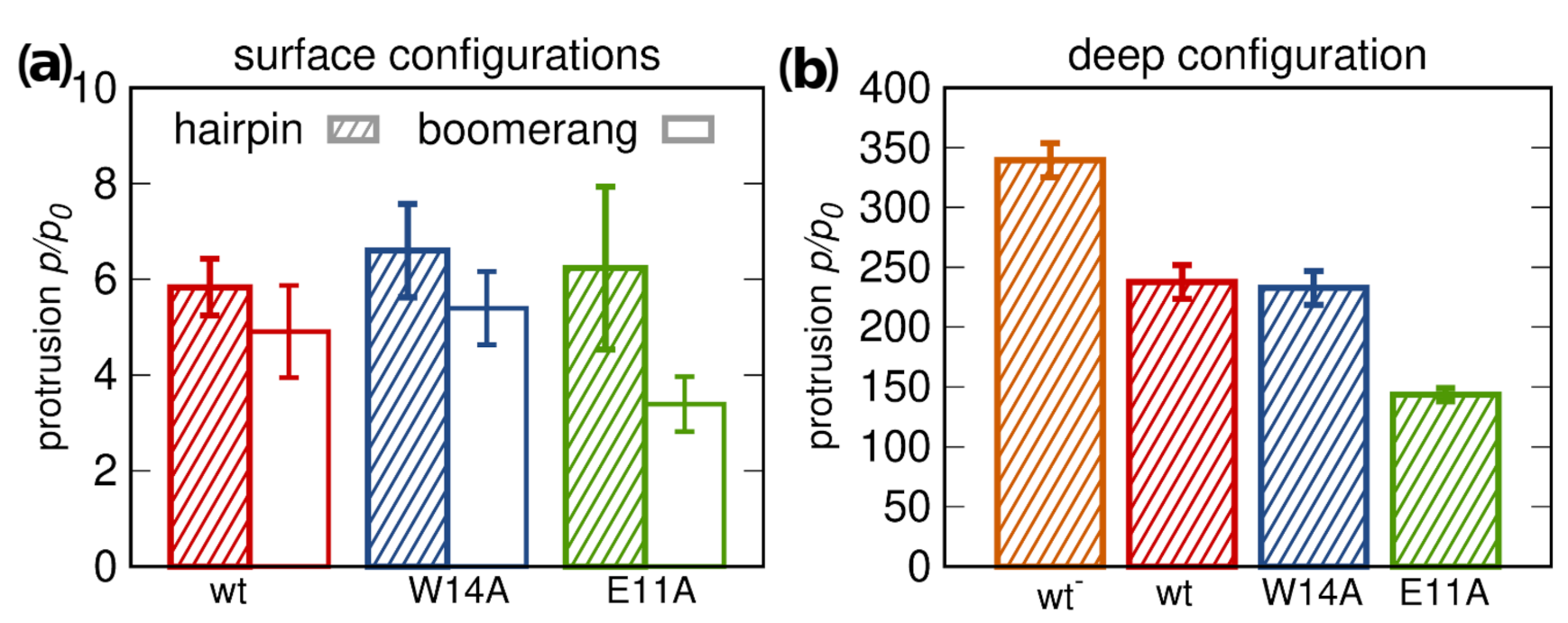
3.5. Membrane Perturbation
3.5.1. Lipid Tail Protrusions
3.5.2. Membrane Water Permeability
3.6. Supervised Insertion
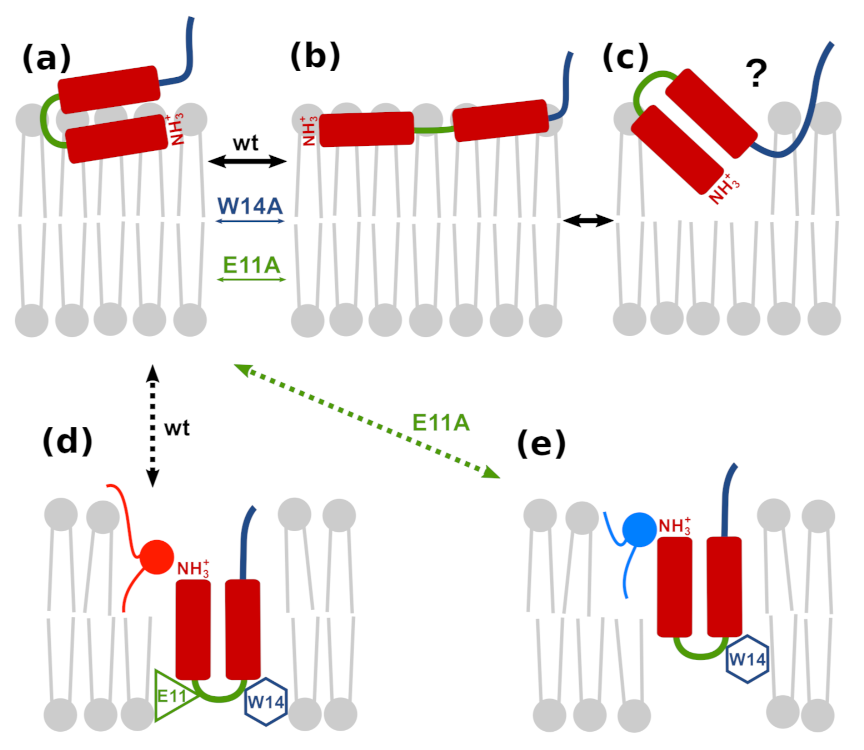
3.7. Possible Insertion Modes
4. Discussion
5. Conclusions
- A model assuming the presence of HAfp exclusively at lipid–water interface is not sufficient to explain mutation-related differences in peptides activity, experimentally estimated helix tilting angle, or peptide-induced membrane water permeability.
- Simulations together with tryptophan fluorescence quenching experiments and the observation of peptide-induced membrane water permeability suggest the existence of membrane-spanning configurations with high membrane-perturbing potential.
- The effect of amino acids mutations on fusogenic activity correlates with peptides ability to achieve and maintain deeply inserted configurations and with the insertion depth of the N-terminal amino group.
- Although surface HAfp binding seems to be the dominant mode, simulations demonstrate the feasibility of spontaneous deep peptide insertions at sufficient rate to promote fusion in experimentally observed time scale.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Durrer, P.; Galli, C.; Hoenke, S.; Corti, C.; Glück, R.; Vorherr, T.; Brunner, J. H+-induced Membrane Insertion of Influenza Virus Hemagglutinin Involves the HA2 Amino-terminal Fusion Peptide but Not the Coiled Coil Region. J. Biol. Chem. 1996, 271, 13417–13421. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.L.; Waring, A.J.; Gordon, L.M.; Hammer, D.A. Area Expansion and Permeation of Phospholipid Membrane Bilayers by Influenza Fusion Peptides and Melittin. Langmuir 1998, 14, 2385–2395. [Google Scholar] [CrossRef]
- Nieva, J.L.; Agirre, A. Are fusion peptides a good model to study viral cell fusion? Biochim. Biophys. Acta Biomembr. 2003, 1614, 104–115. [Google Scholar] [CrossRef]
- Lear, J.D.; DeGrado, W.F. Membrane binding and conformational properties of peptides representing the NH2 terminus of influenza HA-2. J. Biol. Chem. 1987, 262, 6500–6505. [Google Scholar] [CrossRef]
- Rafalski, M.; Rockwell, A.; Lear, J.D.; Grado, W.F.; Wilschut, J.; Ortiz, A.; Ginkel, L.C.; Ortiz, A.; Ginkel, L.C. Membrane Fusion Activity of the Influenza Virus Hemagglutinin: Interaction of HA2 N-Terminal Peptides with Phospholipid Vesicles. Biochemistry 1991, 30, 10211–10220. [Google Scholar] [CrossRef]
- Cross, K.J.; Langley, W.A.; Russell, R.J.; Skehel, J.J.; Steinhauer, D.A. Composition and functions of the influenza fusion peptide. Protein Pept. Lett. 2009, 16, 766–778. [Google Scholar] [CrossRef]
- Martin, I. Structure and Topology of the Influenza Virus Fusion Peptide in Lipid Bilayers. J. Biol. Chem. 1995, 270, 27606–27614. [Google Scholar] [CrossRef]
- Han, X.; Bushweller, J.H.; Cafiso, D.S.; Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001, 8, 715–720. [Google Scholar] [CrossRef]
- Lorieau, J.L.; Louis, J.M.; Bax, A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl. Acad. Sci. USA 2010, 107, 11341–11346. [Google Scholar] [CrossRef]
- Lorieau, J.L.; Louis, J.M.; Bax, A. The impact of influenza hemagglutinin fusion peptide length and viral subtype on its structure and dynamics. Biopolymers 2013, 99, 189–195. [Google Scholar] [CrossRef]
- Tamm, L.K.; Han, X.; Li, Y.; Lai, A.L. Structure and function of membrane fusion peptides. Biopolym. Pept. Sci. Sect. 2002, 66, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.L.; Park, H.; White, J.M.; Tamm, L.K. Fusion Peptide of Influenza Hemagglutinin Requires a Fixed Angle Boomerang Structure for Activity. J. Biol. Chem. 2006, 281, 5760–5770. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A Comprehensive Comparison of Transmembrane Domains Reveals Organelle-Specific Properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef]
- Smrt, S.T.; Draney, A.W.; Lorieau, J.L. The influenza hemagglutinin fusion domain is an amphipathic helical hairpin that functions by inducing membrane curvature. J. Biol. Chem. 2015, 290, 228–238. [Google Scholar] [CrossRef]
- Gray, C.; Tatulian, S.A.; Wharton, S.A.; Tamm, L.K. Effect of the N-terminal glycine on the secondary structure, orientation, and interaction of the influenza hemagglutinin fusion peptide with lipid bilayers. Biophys. J. 1996, 70, 2275–2286. [Google Scholar] [CrossRef]
- Han, X.; Steinhauer, D.A.; Wharton, S.A.; Tamm, L.K. Interaction of mutant influenza virus hemagglutinin fusion peptides with lipid bilayers: Probing the role of hydrophobic residue size in the central region of the fusion peptide. Biochemistry 1999, 38, 15052–15059. [Google Scholar] [CrossRef]
- Victor, B.L.; Lousa, D.; Antunes, J.M.; Soares, C.M. Self-Assembly Molecular Dynamics Simulations Shed Light into the Interaction of the Influenza Fusion Peptide with a Membrane Bilayer. J. Chem. Inf. Model. 2015, 55, 795–805. [Google Scholar] [CrossRef]
- Worch, R.; Krupa, J.; Filipek, A.; Szymaniec, A.; Setny, P. Three conserved C-terminal residues of influenza fusion peptide alter its behavior at the membrane interface. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 97–105. [Google Scholar] [CrossRef]
- Worch, R.; Dudek, A.; Krupa, J.; Szymaniec, A.; Setny, P. Charged n-terminus of influenza fusion peptide facilitates membrane fusion. Int. J. Mol. Sci. 2018, 19, 578. [Google Scholar] [CrossRef] [PubMed]
- Lousa, D.; Pinto, A.R.; Campos, S.R.; Baptista, A.M.; Veiga, A.S.; Castanho, M.A.; Soares, C.M. Effect of pH on the influenza fusion peptide properties unveiled by constant-pH molecular dynamics simulations combined with experiment. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef]
- Larsson, P.; Kasson, P.M. Lipid Tail Protrusion in Simulations Predicts Fusogenic Activity of Influenza Fusion Peptide Mutants and Conformational Models. PLoS Comput. Biol. 2013, 9, e1002950. [Google Scholar] [CrossRef] [PubMed]
- Légaré, S.; Lagüe, P. The influenza fusion peptide promotes lipid polar head intrusion through hydrogen bonding with phosphates and N-terminal membrane insertion depth. Proteins 2014, 82, 2118–2127. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, C.L.; Herrmann, A. Bilayer conformation of fusion peptide of influenza virus hemagglutinin: A molecular dynamics simulation study. Biophys. J. 2004, 87, 14–22. [Google Scholar] [CrossRef]
- Lagüe, P.; Roux, B.; Pastor, R.W. Molecular dynamics simulations of the influenza hemagglutinin fusion peptide in micelles and bilayers: Conformational analysis of peptide and lipids. J. Mol. Biol. 2005, 354, 1129–1141. [Google Scholar] [CrossRef]
- Fuhrmans, M.; Marrink, S.J. Molecular View of the Role of Fusion Peptides in Promoting Positive Membrane Curvature. J. Am. Chem. Soc. 2012, 134, 1543–1552. [Google Scholar] [CrossRef]
- Chakraborty, H.; Lentz, B.R.; Kombrabail, M.; Krishnamoorthy, G.; Chattopadhyay, A. Depth-Dependent Membrane Ordering by Hemagglutinin Fusion Peptide Promotes Fusion. J. Phys. Chem. B 2017, 121, 1640–1648. [Google Scholar] [CrossRef]
- Risselada, H.J.; Marelli, G.; Fuhrmans, M.; Smirnova, Y.G.; Grubmüller, H.; Marrink, S.j.; Müller, M. Line-tension controlled mechanism for influenza fusion. PLoS ONE 2012, 7, 16–20. [Google Scholar] [CrossRef]
- Tahir, M.A.; Van Lehn, R.C.; Choi, S.H.; Alexander-Katz, A. Solvent-exposed lipid tail protrusions depend on lipid membrane composition and curvature. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1207–1215. [Google Scholar] [CrossRef]
- Pabis, A.; Rawle, R.J.; Kasson, P.M. Influenza hemagglutinin drives viral entry via two sequential intramembrane mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 7200–7207. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páall, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Seibert, M.M. Protein Folding Kinetics and Thermodynamics from Atomistic Simulations. Phys. Rev. Lett. 2006, 96, 238102–238104. [Google Scholar] [CrossRef] [PubMed]
- Souaille, M.; Roux, B. Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Bussi, G.; Tribello, G.A. Analyzing and Biasing Simulations with PLUMED. Methods Mol. Biol. 2019, 2022, 529–578. [Google Scholar] [CrossRef]
- Ladokhin, A.S. Distribution analysis of depth-dependent fluorescence quenching in membranes: A practical guide. Methods Enzymol. 1997, 278, 462–473. [Google Scholar] [CrossRef]
- Marrink, S.J.; Berendsen, H.J.C. Simulation of water transport through a lipid membrane. J. Phys. Chem. 1994, 98, 4155–4168. [Google Scholar] [CrossRef]
- Mathai, J.C.; Tristram-Nagle, S.; Nagle, J.F.; Zeidel, M.L. Structural Determinants of Water Permeability through the Lipid Membrane. J. Gen. Physiol. 2008, 131, 69–76. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Jayasinghe, S.; White, S.H. How to Measure and Analyze Tryptophan Fluorescence in Membranes Properly, and Why Bother? Anal. Biochem. 2000, 285, 235–245. [Google Scholar] [CrossRef]
- Lorieau, J.L.; Louis, J.M.; Schwieters, C.D.; Bax, A. pH-triggered, activated-state conformations of the influenza hemagglutinin fusion peptide revealed by NMR. Proc. Natl. Acad. Sci. USA 2012, 109, 19994–19999. [Google Scholar] [CrossRef]
- Ghosh, U.; Xie, L.; Jia, L.; Liang, S.; Weliky, D.P. Closed and Semiclosed Interhelical Structures in Membrane vs Closed and Open Structures in Detergent for the Influenza Virus Hemagglutinin Fusion Peptide and Correlation of Hydrophobic Surface Area with Fusion Catalysis. J. Am. Chem. Soc. 2015, 137, 7548–7551. [Google Scholar] [CrossRef]
- Tamm, L.K.; Tatulian, S.A. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 1997, 30, 365–429. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Cheng, S.F.; Huang, W.N.; Trivedi, V.D.; Veeramuthu, B.; Kantchev, A.B.; Wu, W.G.; Chang, D.K. Effects of alterations of the amino-terminal glycine of influenza hemagglutinin fusion peptide on its structure, organization and membrane interactions. Biochim. Biophys. Acta Biomembr. 2003, 1612, 41–51. [Google Scholar] [CrossRef]
- Han, X.; Tamm, L.K. A host-guest system to study structure-function relationships of membrane fusion peptides. Proc. Natl. Acad. Sci. USA 2000, 97, 13097–13102. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S.; Holloway, P.W. Fluorescence of membrane-bound tryptophan octyl ester: A model for studying intrinsic fluorescence of protein-membrane interactions. Biophys. J. 1995, 69, 506–517. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | ||||||||
|---|---|---|---|---|---|---|---|---|
| wt | ||||||||
| W14A | ||||||||
| E11A | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Worch, R.; Dudek, A.; Borkowska, P.; Setny, P. Transient Excursions to Membrane Core as Determinants of Influenza Virus Fusion Peptide Activity. Int. J. Mol. Sci. 2021, 22, 5301. https://doi.org/10.3390/ijms22105301
Worch R, Dudek A, Borkowska P, Setny P. Transient Excursions to Membrane Core as Determinants of Influenza Virus Fusion Peptide Activity. International Journal of Molecular Sciences. 2021; 22(10):5301. https://doi.org/10.3390/ijms22105301
Chicago/Turabian StyleWorch, Remigiusz, Anita Dudek, Paulina Borkowska, and Piotr Setny. 2021. "Transient Excursions to Membrane Core as Determinants of Influenza Virus Fusion Peptide Activity" International Journal of Molecular Sciences 22, no. 10: 5301. https://doi.org/10.3390/ijms22105301
APA StyleWorch, R., Dudek, A., Borkowska, P., & Setny, P. (2021). Transient Excursions to Membrane Core as Determinants of Influenza Virus Fusion Peptide Activity. International Journal of Molecular Sciences, 22(10), 5301. https://doi.org/10.3390/ijms22105301