
Liver Lipids of Patients with Hepatitis B and C and Associated Hepatocellular Carcinoma


 , ,
, ,  , and
, and 
Abstract
1. Introduction
2. Results
2.1. Lipidomic Analysis of Tumor and Adjacent Tissues of Patients with Chronic HBV and HCV Infections
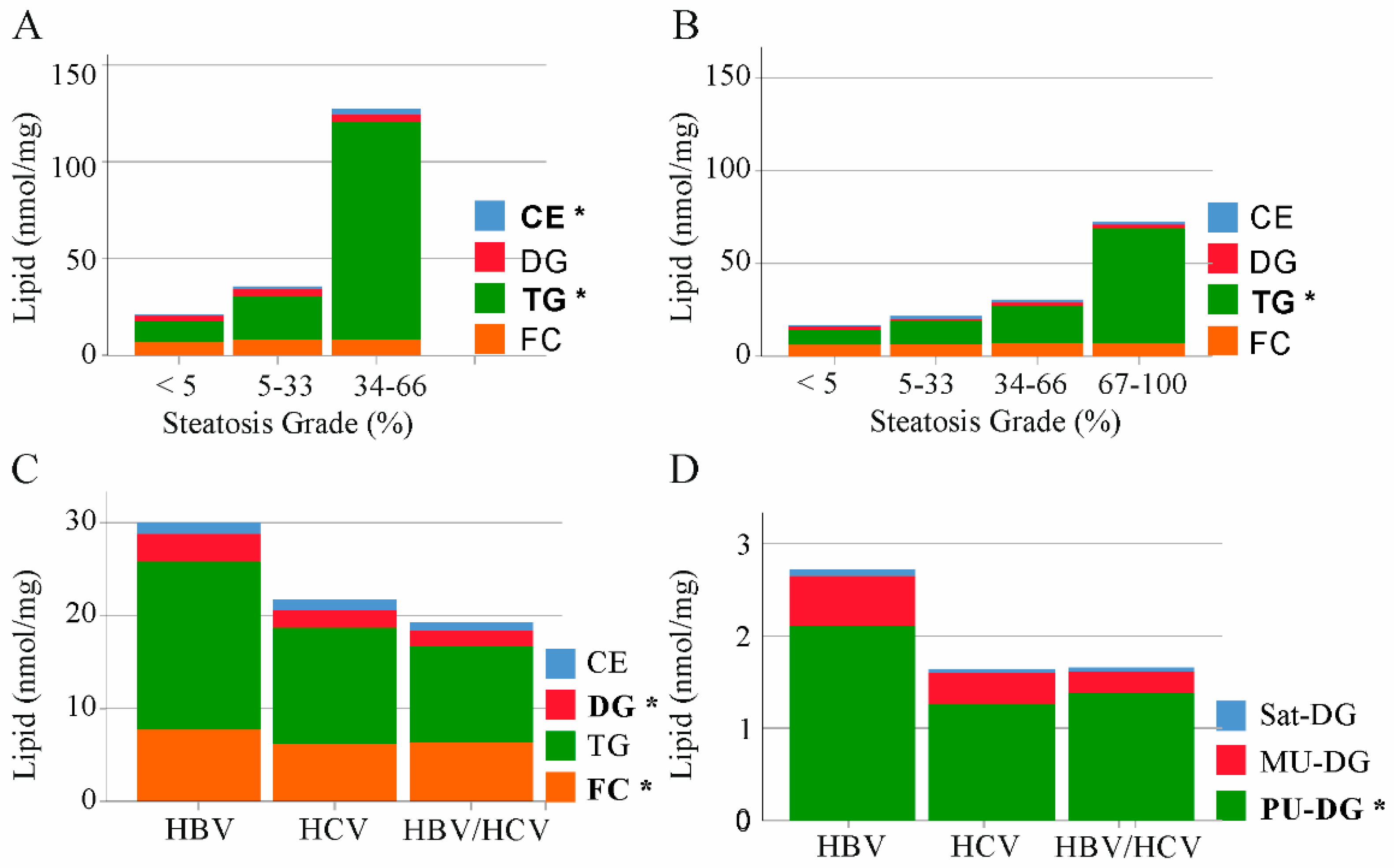
2.2. Triacylglycerols and Cholesterol in Non-Tumor Tissues of HBV and HCV Infected Patients
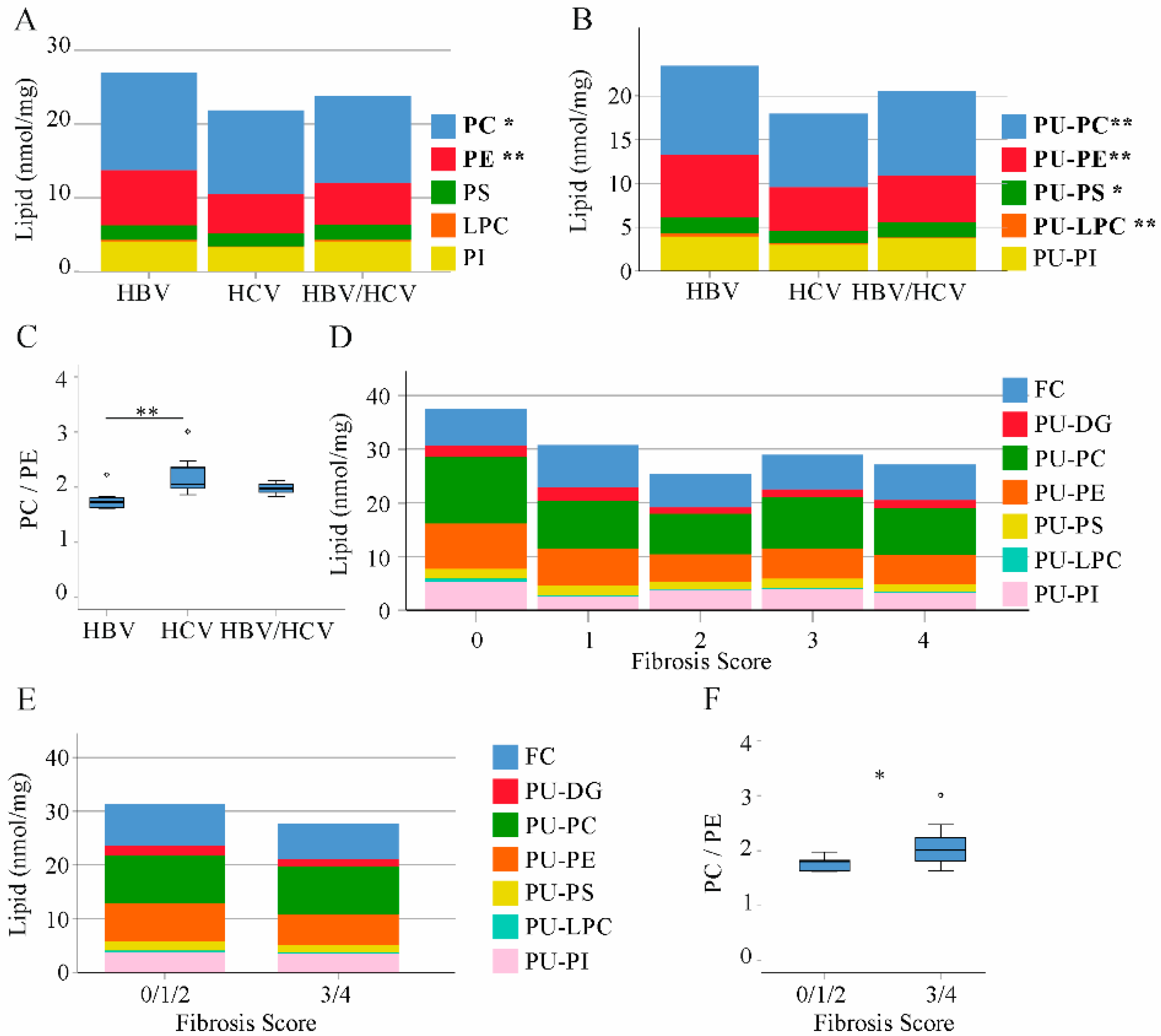
2.3. Phospholipids in Non-Tumor Tissues of HBV and HCV Infected Patients
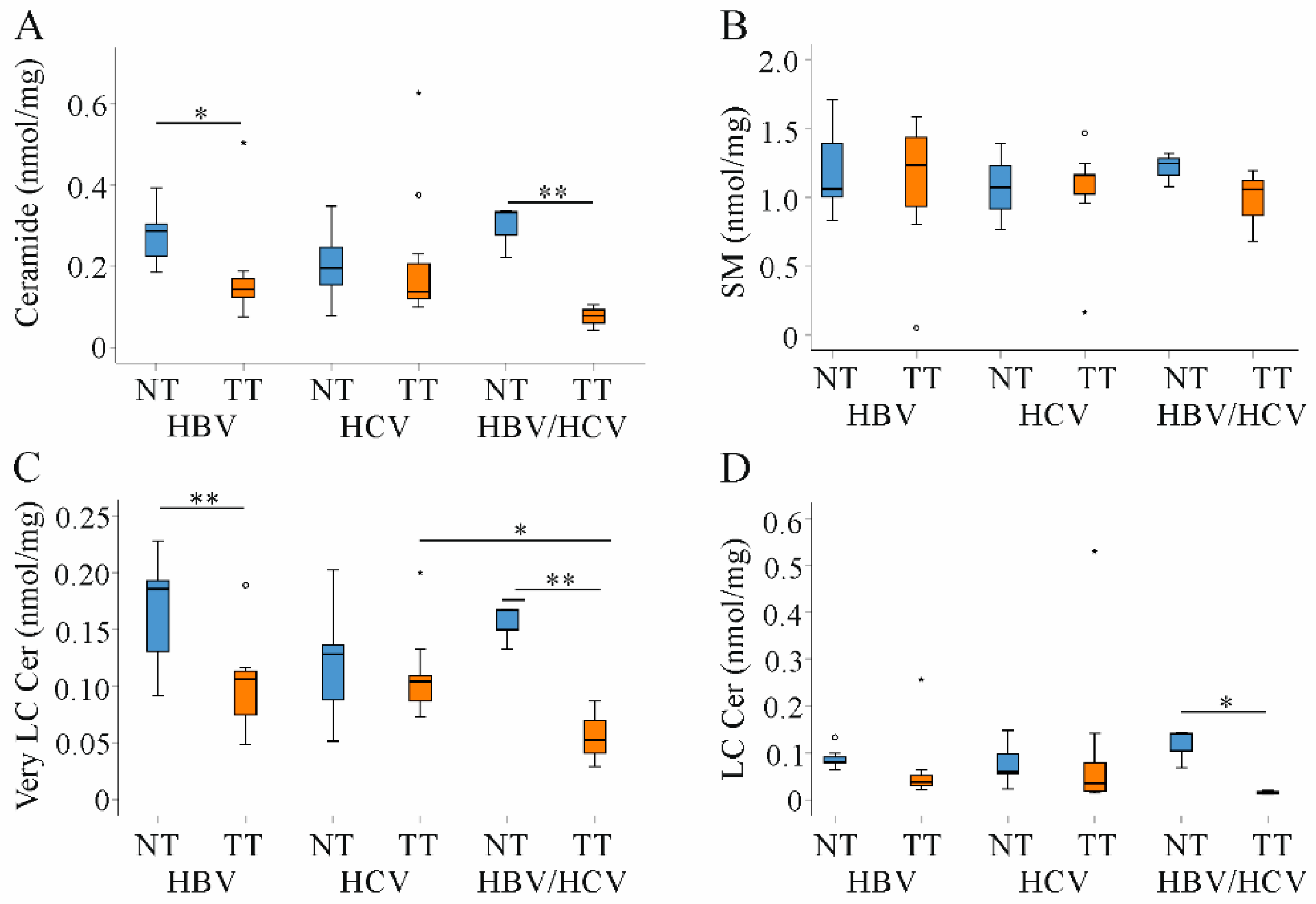
2.4. Ceramide and Sphingomyelin in Non-Tumor and Tumor Tissues of HBV and HCV Infected Patients
2.5. Cholesterol, Tri- and Diacylglycerides in Tumor Tissues of HBV and HCV Infected Patients
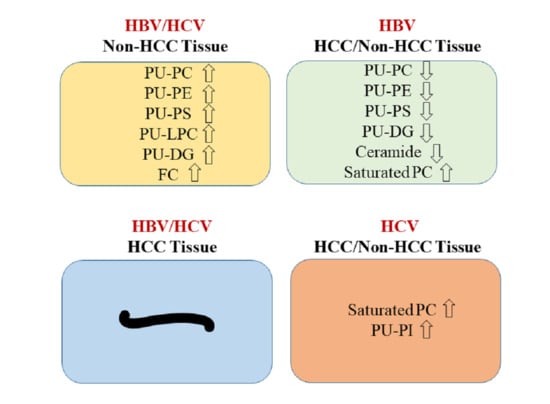
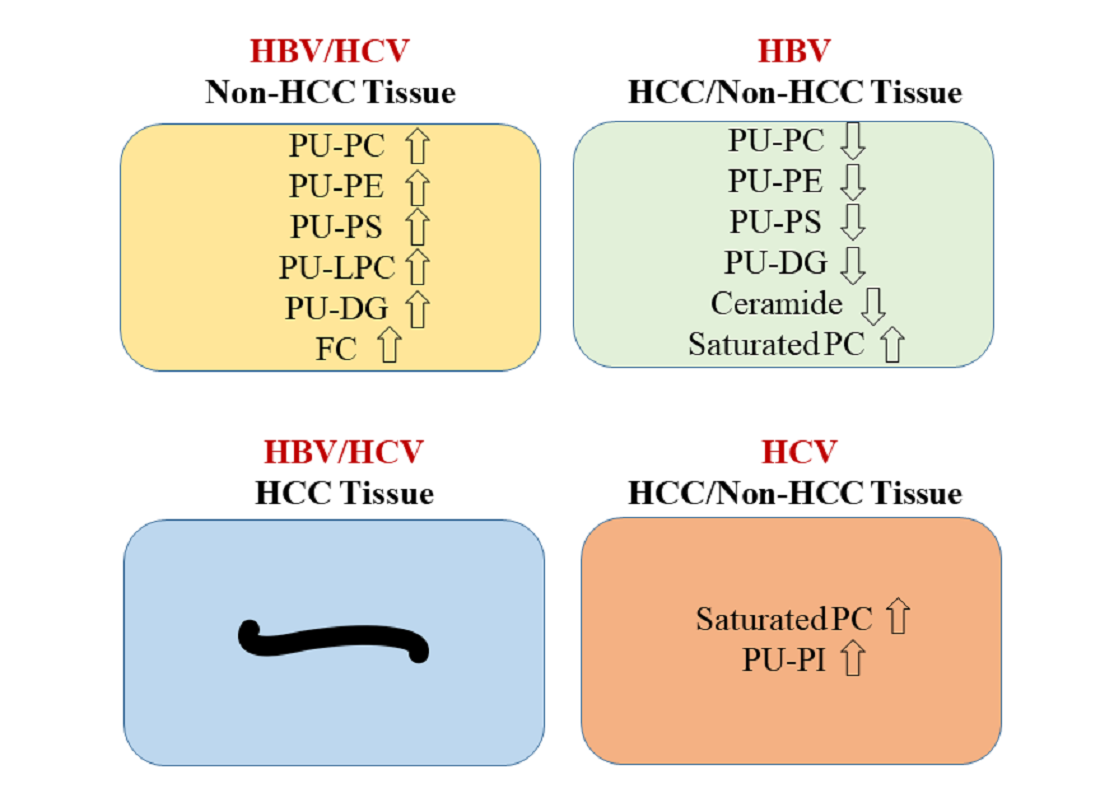
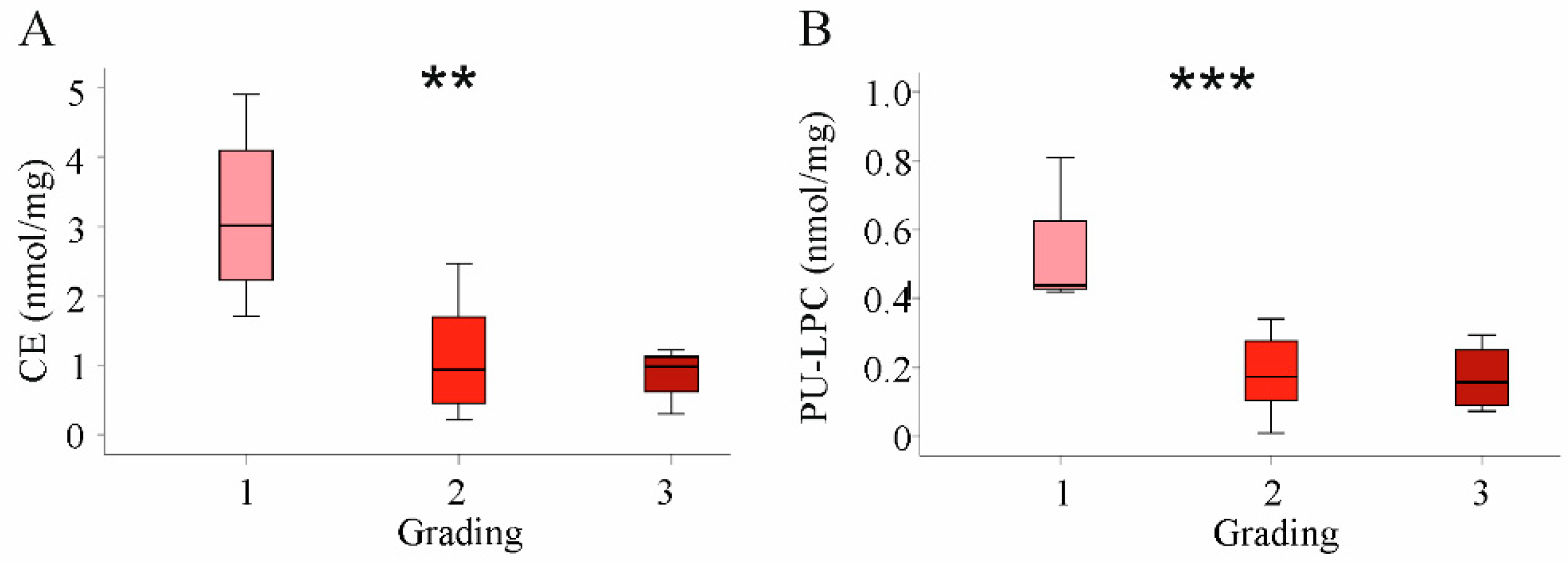
2.6. Decreased PU-Phospholipids in HBV Tumors
2.7. Correlation of Lipids in HCC Tissues with Tumor Grading and Stage
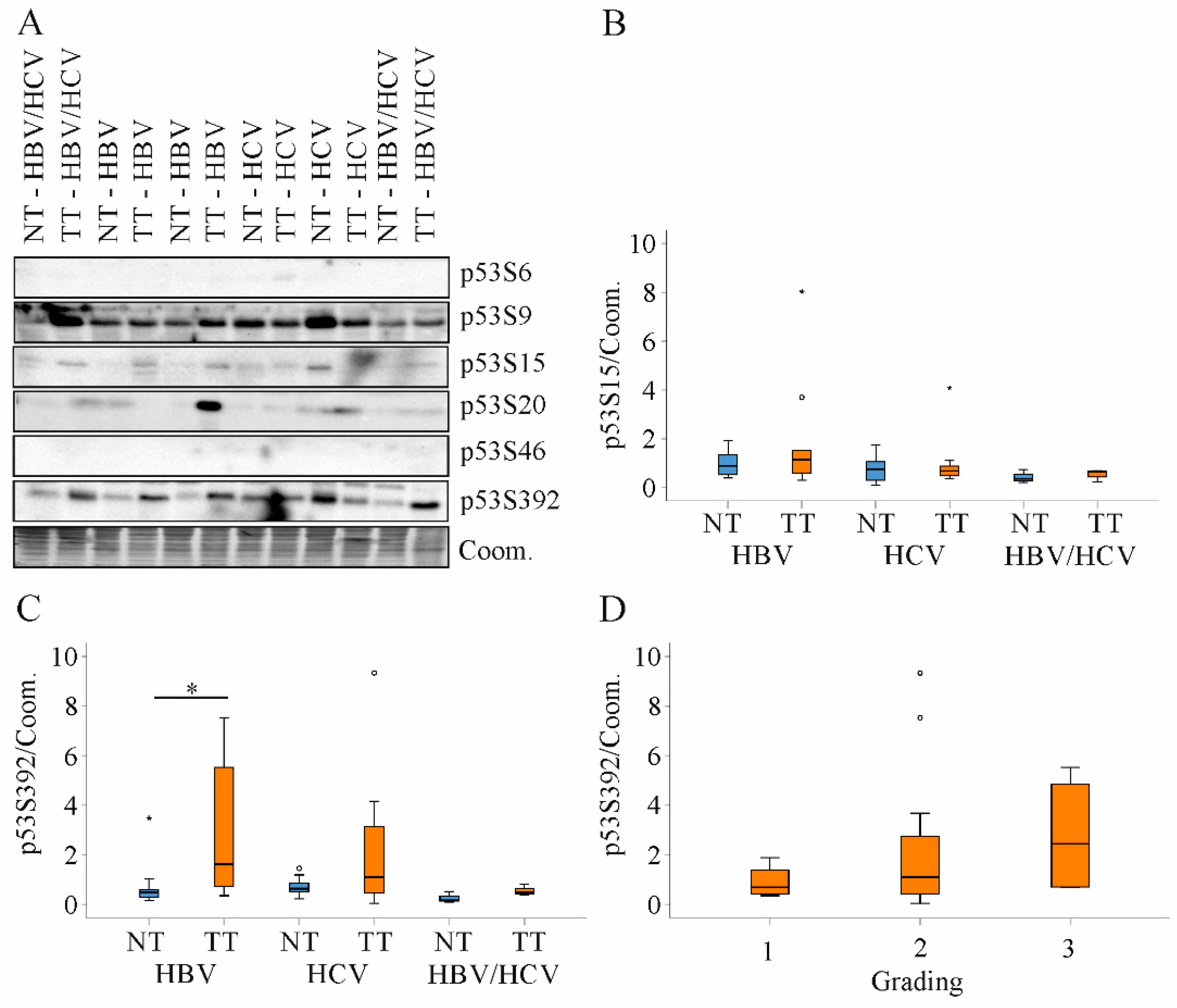
2.8. Expression of p53 Protein in HBV and HCV Infected Patients
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Lipidomics
4.2.1. Internal Standards
4.2.2. Lipid Extraction
4.2.3. Lipid Analysis
4.3. Immunoblot Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Song, K.H.; Koo, I.; Lee, K.H.; Suh, K.S.; Kim, B.Y. Comparison of pathways associated with hepatitis B- and C-infected hepatocellular carcinoma using pathway-based class discrimination method. Genomics 2012, 99, 347–354. [Google Scholar] [CrossRef][Green Version]
- Zampino, R.; Pisaturo, M.A.; Cirillo, G.; Marrone, A.; Macera, M.; Rinaldi, L.; Stanzione, M.; Durante-Mangoni, E.; Gentile, I.; Sagnelli, E.; et al. Hepatocellular carcinoma in chronic HBV-HCV co-infection is correlated to fibrosis and disease duration. Ann. Hepatol. 2015, 14, 75–82. [Google Scholar] [CrossRef]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Cheng, P.N.; Kao, J.H. Systematic review: Chronic viral hepatitis and metabolic derangement. Aliment Pharmacol. Ther. 2020, 51, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Feder, S.; Wiest, R.; Weiss, T.S.; Aslanidis, C.; Schacherer, D.; Krautbauer, S.; Liebisch, G.; Buechler, C. Proprotein convertase subtilisin/kexin type 9 (PCSK9) levels are not associated with severity of liver disease and are inversely related to cholesterol in a cohort of thirty eight patients with liver cirrhosis. Lipids Health Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhu, P.; Liang, Y.; Yin, W.G.; Xiao, J.H. Hepatitis B virus induces expression of cholesterol metabolism-related genes via TLR2 in HepG2 cells. World J. Gastroenterol. 2013, 19, 2262–2269. [Google Scholar] [CrossRef]
- Grammatikos, G.; Ferreiros, N.; Bon, D.; Schwalm, S.; Dietz, J.; Berkowski, C.; Fitting, D.; Herrmann, E.; Zeuzem, S.; Sarrazin, C.; et al. Variations in serum sphingolipid levels associate with liver fibrosis progression and poor treatment outcome in hepatitis C virus but not hepatitis B virus infection. Hepatology 2015, 61, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.J.; Qu, F.; Li, J.F.; Zhao, J.; Zhang, J.Y.; Liu, M.; Ren, F.; Chen, Y.; Zhang, J.L.; Duan, Z.P. Serum sphingomyelin has potential to reflect hepatic injury in chronic hepatitis B virus infection. Int. J. Infect. Dis. 2015, 33, 149–155. [Google Scholar] [CrossRef]
- Huang, Q.; Lei, H.; Ding, L.; Wang, Y. Stimulated phospholipid synthesis is key for hepatitis B virus replications. Sci. Rep. 2019, 9, 12989. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Okamoto, K.; Aoki, M.; Kato, H.; Katsume, A.; Ohta, A.; Tsukuda, T.; Shimma, N.; Aoki, Y.; Arisawa, M.; et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat. Chem. Biol. 2005, 1, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J. Lipid Res. 2009, 50, 574–585. [Google Scholar] [CrossRef]
- Wang, S.B.; Ma, Y.Y.; Chen, X.Y.; Zhao, Y.Y.; Mou, X.Z. Ceramide-Graphene Oxide Nanoparticles Enhance Cytotoxicity and Decrease HCC Xenograft Development: A Novel Approach for Targeted Cancer Therapy. Front. Pharmacol. 2019, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1412. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Eckstein, J.; Heucke, N.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhutter, H.G. Characterization of Lipid and Lipid Droplet Metabolism in Human HCC. Cells 2019, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; De Kock, M.; van Schalkwyk, D.J.; Swanevelder, S.; Kew, M.C.; Gelderblom, W.C. Altered lipid profile, oxidative status and hepatitis B virus interactions in human hepatocellular carcinoma. Prostaglandins Leukot Essent Fat. Acids 2009, 81, 391–399. [Google Scholar] [CrossRef]
- Krautbauer, S.; Meier, E.M.; Rein-Fischboeck, L.; Pohl, R.; Weiss, T.S.; Sigruener, A.; Aslanidis, C.; Liebisch, G.; Buechler, C. Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochim. Biophys. Acta 2016, 1861, 1767–1774. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant Lipid Metabolism in Hepatocellular Carcinoma Revealed by Liver Lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef]
- Lin, L.; Ding, Y.; Wang, Y.; Wang, Z.; Yin, X.; Yan, G.; Zhang, L.; Yang, P.; Shen, H. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology 2017, 66, 432–448. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Huang, C.; Li, N.; Zou, L.; Chia, S.E.; Chen, S.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Comparison of hepatic and serum lipid signatures in hepatocellular carcinoma patients leads to the discovery of diagnostic and prognostic biomarkers. Oncotarget 2018, 9, 5032–5043. [Google Scholar] [CrossRef] [PubMed]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta 2015, 1852, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Abenavoli, L.; Masarone, M.; Peta, V.; Milic, N.; Kobyliak, N.; Rouabhia, S.; Persico, M. Insulin resistance and liver steatosis in chronic hepatitis C infection genotype 3. World J. Gastroenterol. 2014, 20, 15233–15240. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Brown, S.H.; Lim, X.Y.; Fiveash, C.E.; Osborne, B.; Bentley, N.L.; Braude, J.P.; Mitchell, T.W.; Coster, A.C.; Don, A.S.; et al. Regulation of glucose homeostasis and insulin action by ceramide acyl-chain length: A beneficial role for very long-chain sphingolipid species. Biochim. Biophys. Acta 2016, 1861, 1828–1839. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef]
- Wang, H.Y.; Quan, C.; Hu, C.; Xie, B.; Du, Y.; Chen, L.; Yang, W.; Yang, L.; Chen, Q.; Shen, B.; et al. A lipidomics study reveals hepatic lipid signatures associating with deficiency of the LDL receptor in a rat model. Biol. Open 2016, 5, 979–986. [Google Scholar] [CrossRef]
- Lambert, J.E.; Bain, V.G.; Ryan, E.A.; Thomson, A.B.; Clandinin, M.T. Elevated lipogenesis and diminished cholesterol synthesis in patients with hepatitis C viral infection compared to healthy humans. Hepatology 2013, 57, 1697–1704. [Google Scholar] [CrossRef]
- Na, T.Y.; Shin, Y.K.; Roh, K.J.; Kang, S.A.; Hong, I.; Oh, S.J.; Seong, J.K.; Park, C.K.; Choi, Y.L.; Lee, M.O. Liver X receptor mediates hepatitis B virus X protein-induced lipogenesis in hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2009, 49, 1122–1131. [Google Scholar] [CrossRef]
- Roingeard, P. Hepatitis C virus diversity and hepatic steatosis. J. Viral. Hepat 2013, 20, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Peschel, G.; Grimm, J.; Gulow, K.; Muller, M.; Buechler, C.; Weigand, K. Chemerin Is a Valuable Biomarker in Patients with HCV Infection and Correlates with Liver Injury. Diagnostics 2020, 10, 974. [Google Scholar] [CrossRef] [PubMed]
- Geier, A. Hepatitis B virus: The “metabolovirus” highjacks cholesterol and bile acid metabolism. Hepatology 2014, 60, 1458–1460. [Google Scholar] [CrossRef]
- Cabre, E.; Gassull, M.A. Polyunsaturated fatty acid deficiency in liver diseases: Pathophysiological and clinical significance. Nutrition 1996, 12, 542–548. [Google Scholar] [CrossRef]
- Enguita, M.; Razquin, N.; Pamplona, R.; Quiroga, J.; Prieto, J.; Fortes, P. The cirrhotic liver is depleted of docosahexaenoic acid (DHA), a key modulator of NF-kappaB and TGFbeta pathways in hepatic stellate cells. Cell Death Dis. 2019, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Sasagawa, T.; Suzuki, K.; Tomioka, K.; Nagai, A.; Niiyama, G.; Kawanaka, M.; Yamada, G.; Okita, M. Vitamin E supplementation increases polyunsaturated fatty acids of RBC membrane in HCV-infected patients. Nutrition 2004, 20, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef]
- Vance, J.E.; Tasseva, G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta 2013, 1831, 543–554. [Google Scholar] [CrossRef]
- Gobeil Odai, K.; O’Dwyer, C.; Steenbergen, R.; Shaw, T.A.; Renner, T.M.; Ghorbani, P.; Rezaaifar, M.; Han, S.; Langlois, M.A.; Crawley, A.M.; et al. In Vitro Hepatitis C Virus Infection and Hepatic Choline Metabolism. Viruses 2020, 12, 108. [Google Scholar] [CrossRef]
- Wong, M.T.; Chen, S.S. Human Choline Kinase-alpha Promotes Hepatitis C Virus RNA Replication through Modulation of Membranous Viral Replication Complex Formation. J. Virol. 2016, 90, 9075–9095. [Google Scholar] [CrossRef]
- Eichmann, T.O.; Lass, A. DAG tales: The multiple faces of diacylglycerol--stereochemistry, metabolism, and signaling. Cell Mol. Life Sci. 2015, 72, 3931–3952. [Google Scholar] [CrossRef] [PubMed]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chretien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB J. 2002, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Munakata, T.; Kohara, M.; Siddiqui, A.; Peers, C.; Harris, M. Hepatitis C virus attenuates mitochondrial lipid beta-oxidation by downregulating mitochondrial trifunctional-protein expression. J. Virol. 2015, 89, 4092–4101. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Zhou, R.F.; Chen, X.L.; Zhou, Z.G.; Zhang, Y.J.; Lan, Q.Y.; Liao, G.C.; Chen, Y.M.; Zhu, H.L. Higher dietary intakes of choline and betaine are associated with a lower risk of primary liver cancer: A case-control study. Sci. Rep. 2017, 7, 679. [Google Scholar] [CrossRef]
- Bi, J.; Ichu, T.A.; Zanca, C.; Yang, H.; Zhang, W.; Gu, Y.; Chowdhry, S.; Reed, A.; Ikegami, S.; Turner, K.M.; et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metab. 2019, 30, 525–538.e8. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, Q. Do hepatitis B virus and hepatitis C virus co-infections increase hepatocellular carcinoma occurrence through synergistically modulating lipogenic gene expression? Hepatol. Res. 2012, 42, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Eggens, I.; Ekstrom, T.J.; Aberg, F. Studies on the biosynthesis of polyisoprenols, cholesterol and ubiquinone in highly differentiated human hepatomas. J. Exp. Pathol. 1990, 71, 219–232. [Google Scholar]
- Goldstein, I.; Ezra, O.; Rivlin, N.; Molchadsky, A.; Madar, S.; Goldfinger, N.; Rotter, V. p53, a novel regulator of lipid metabolism pathways. J. Hepatol. 2012, 56, 656–662. [Google Scholar] [CrossRef]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef]
- Muller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, two, three--p53, p63, p73 and chemosensitivity. Drug Resist Updat. 2006, 9, 288–306. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; D’Errico, A.; Paterlini, P.; Grigioni, W.; May, E.; Debuire, B. p53 protein accumulation in European hepatocellular carcinoma is not always dependent on p53 gene mutation. Gastroenterology 1995, 108, 1176–1182. [Google Scholar] [CrossRef]
- Bist, A.; Fielding, C.J.; Fielding, P.E. p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry 2000, 39, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.L.; Meek, D.W. Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell. Signal. 2010, 22, 564–571. [Google Scholar] [CrossRef]
- Yang, T.; Choi, Y.; Joh, J.W.; Cho, S.K.; Kim, D.S.; Park, S.G. Phosphorylation of p53 Serine 15 Is a Predictor of Survival for Patients with Hepatocellular Carcinoma. Can J. Gastroenterol. Hepatol. 2019, 2019, 9015453. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Ten Hove, M.; Pater, L.; Storm, G.; Weiskirchen, S.; Weiskirchen, R.; Lammers, T.; Bansal, R. The hepatic lipidome: From basic science to clinical translation. Adv. Drug Deliv. Rev. 2020, 159, 180–197. [Google Scholar] [CrossRef]
- Haberl, E.M.; Feder, S.; Pohl, R.; Rein-Fischboeck, L.; Durholz, K.; Eichelberger, L.; Wanninger, J.; Weiss, T.S.; Buechler, C. Chemerin Is Induced in Non-Alcoholic Fatty Liver Disease and Hepatitis B-Related Hepatocellular Carcinoma. Cancers 2020, 12, 2967. [Google Scholar] [CrossRef]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Kofeler, H.; et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Liebisch, G.; Lieser, B.; Rathenberg, J.; Drobnik, W.; Schmitz, G. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope correction algorithm. Biochim. Biophys. Acta 2004, 1686, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Binder, M.; Schifferer, R.; Langmann, T.; Schulz, B.; Schmitz, G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta 2006, 1761, 121–128. [Google Scholar] [CrossRef]
- Liebisch, G.; Drobnik, W.; Lieser, B.; Schmitz, G. High-throughput quantification of lysophosphatidylcholine by electrospray ionization tandem mass spectrometry. Clin. Chem. 2002, 48, 2217–2224. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Drobnik, W.; Reil, M.; Trumbach, B.; Arnecke, R.; Olgemoller, B.; Roscher, A.; Schmitz, G. Quantitative measurement of different ceramide species from crude cellular extracts by electrospray ionization tandem mass spectrometry (ESI-MS/MS). J. Lipid Res. 1999, 40, 1539–1546. [Google Scholar] [CrossRef]
- Höring, M.; Ejsing, C.S.; Krautbauer, S.; Ertl, V.M.; Burkhardt, R.; Liebisch, G. Accurate quantification of lipid species affected by isobaric overlap in Fourier-Transform mass spectrometry. J. Lipid Res. 2021, 100050. [Google Scholar] [CrossRef]
- Höring, M.; Ekroos, K.; Baker, P.R.S.; Connell, L.; Stadler, S.C.; Burkhardt, R.; Liebisch, G. Correction of Isobaric Overlap Resulting from Sodiated Ions in Lipidomics. Anal. Chem. 2020, 92, 10966–10970. [Google Scholar] [CrossRef] [PubMed]
- Horing, M.; Ejsing, C.S.; Hermansson, M.; Liebisch, G. Quantification of Cholesterol and Cholesteryl Ester by Direct Flow Injection High Resolution FTMS Utilizing Species-Specific Response Factors. Anal. Chem. 2019. [Google Scholar] [CrossRef]
- Husen, P.; Tarasov, K.; Katafiasz, M.; Sokol, E.; Vogt, J.; Baumgart, J.; Nitsch, R.; Ekroos, K.; Ejsing, C.S. Analysis of lipid experiments (ALEX): A software framework for analysis of high-resolution shotgun lipidomics data. PLoS ONE 2013, 8, e79736. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, J.; Neumeier, M.; Bauer, S.; Weiss, T.S.; Eisinger, K.; Walter, R.; Dorn, C.; Hellerbrand, C.; Schaffler, A.; Buechler, C. Adiponectin induces the transforming growth factor decoy receptor BAMBI in human hepatocytes. FEBS Lett. 2011, 585, 1338–1344. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HBV | HCV | HBV/HCV | ||||
|---|---|---|---|---|---|---|
| NT | TT | NT | TT | NT | TT | |
| Sat. PC | 0.23 (0.18–0.32) | 0.49 * (0.15–1.00) | 0.28 (0.18–0.55) | 0.43 * (0.11–1.08) | 0.29 (0.26–0.34) | 0.32 (0.12–0.70) |
| PU-PC | 10.2 (6.5–14.7) | 5.9 *** (3.2–8.6) | 8.4 (3.3–10.2) | 7.6 (0.6–16.8) | 9.6 (7.6–11.1) | 7.5 (1.7–10.6) |
| PU-PE | 7.2 (3.8–9.9) | 4.7 ** (0.1–8.0) | 5.0 (1.7–6.9) | 6.4 (0.1–10.6) | 5.4 (5.1–7.2) | 6.8 (1.1–7.1) |
| PC/PE | 1.7 (1.6–2.2) | 2.1 (1.5–74.6) | 2.1 (1.7–3.0) | 1.9 (1.7–12.4) | 2.0 (1.8–2.1) | 2.1 (1.9–2.1) |
| Sat. PI | 0.02 (0.01–0.03) | 0.01 (0.00–0.03) | 0.02 (0.01–0.05) | 0.03 (0.00–0.07) | 0.02 (0.02–0.02) | 0.02 (0.01–0.10) |
| PU-PI | 3.9 (2.3–5.6) | 4.1 (0.1–6.4) | 3.0 (1.4–4.8) | 4.7 * (0.1–9.6) | 3.8 (3.5–4.2) | 5.6 (0.8–7.8) |
| PU-PS | 1.8 (1.1–2.2) | 1.2 *** (0.1–1.9) | 1.4 (0.7–1.8) | 1.5 (0.1–2.2) | 1.7 (1.4–1.8) | 1.2 (0.6–1.6) |
| Sat. LPC | 0.07 (0.04–0.10) | 0.08 (0.04–5.81) | 0.06 (0.02–0.26) | 0.06 (0.03–0.10) | 0.08 (0.07–0.18) | 0.04 (0.02–0.05) |
| PU-LPC | 0.40 (0.23–0.86) | 0.38 (0.21–0.81) | 0.17 (0.06–0.31) | 0.14 (0.07–0.29) | 0.06 (0.05–0.13) | 0.03 (0.01–0.05) |
| p53S9 | p53S15 | P53S20 | p53S392 | |
|---|---|---|---|---|
| p53 | −0.018 | −0.052 | 0.327 | 0.347 |
| p53S9 | 0.916 *** | 0.685 * | 0.685 * | |
| p53S15 | 0.916 *** | 0.755 ** | 0.598 ** | |
| p53S20 | 0.685 * | 0.755 ** | 0.797 ** | |
| p53S392 | 0.587 * | 0.598 ** | 0.797 ** | |
| Free cholesterol | 0.073 | 0.128 | 0.027 | −0.129 |
| Grading | −0.194 | −0.033 | −0.065 | 0.222 |
| T-stage | −0.130 | −0.072 | −0.145 | 0.063 |
| HBV | HCV | HBV/HCV | |
|---|---|---|---|
| Number | 10 | 11 | 3 |
| Sex (male/female) | 8/2 | 8/3 | 2/1 |
| Type 2 diabetes | 2 | 3 | 0 |
| Age (years) | 60 (35–78) | 54 (48–71) | 61 (54–76) |
| BMI (kg/m²) | 24.5 (18.7–29.4) | 25.2 (18.8–28.7) | 26.2 (22.4 –30.0) |
| AST (U/L) | 40 (19–103) * | 75 (39–151) | 95 (78–200) * |
| ALT (U/L) | 40 (24–123) * | 60 (27–145) | 134 (66–167) * |
| Bilirubin (mg/dL) | 0.7 (0.3–1.6) | 0.7 (0.4–3.7) | 0.5 (0.5–0.8) |
| GGT (U/L) | 97 (34–200)9,* | 271 (55–582) * | 102 (84–240) |
| Steatosis Grade 0/1/2/3/nd | 7/1/2/0 | 6/2/1/1/1 | 2/0/0/1/0 |
| Fibrosis Stage 0/1/2/3/4 | 2/2/0/3/3 * | 0/0/0/3/8 * | 0/0/1/1/1 |
| Grading G1/G2/nd | 1/4/5 | 0/8/3 | 0/1/2 |
| Staging T1/T2/T3/nd | 2/3/1/4 | 3/3/2/3 | 1/0/1/1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haberl, E.M.; Weiss, T.S.; Peschel, G.; Weigand, K.; Köhler, N.; Pauling, J.K.; Wenzel, J.J.; Höring, M.; Krautbauer, S.; Liebisch, G.; et al. Liver Lipids of Patients with Hepatitis B and C and Associated Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 5297. https://doi.org/10.3390/ijms22105297
Haberl EM, Weiss TS, Peschel G, Weigand K, Köhler N, Pauling JK, Wenzel JJ, Höring M, Krautbauer S, Liebisch G, et al. Liver Lipids of Patients with Hepatitis B and C and Associated Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(10):5297. https://doi.org/10.3390/ijms22105297
Chicago/Turabian StyleHaberl, Elisabeth M., Thomas S. Weiss, Georg Peschel, Kilian Weigand, Nikolai Köhler, Josch K. Pauling, Jürgen J. Wenzel, Marcus Höring, Sabrina Krautbauer, Gerhard Liebisch, and et al. 2021. "Liver Lipids of Patients with Hepatitis B and C and Associated Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 10: 5297. https://doi.org/10.3390/ijms22105297
APA StyleHaberl, E. M., Weiss, T. S., Peschel, G., Weigand, K., Köhler, N., Pauling, J. K., Wenzel, J. J., Höring, M., Krautbauer, S., Liebisch, G., & Buechler, C. (2021). Liver Lipids of Patients with Hepatitis B and C and Associated Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(10), 5297. https://doi.org/10.3390/ijms22105297