
The Multifunctional Role of EMP3 in the Regulation of Membrane Receptors Associated with IDH-Wild-Type Glioblastoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. EMP3 Biology
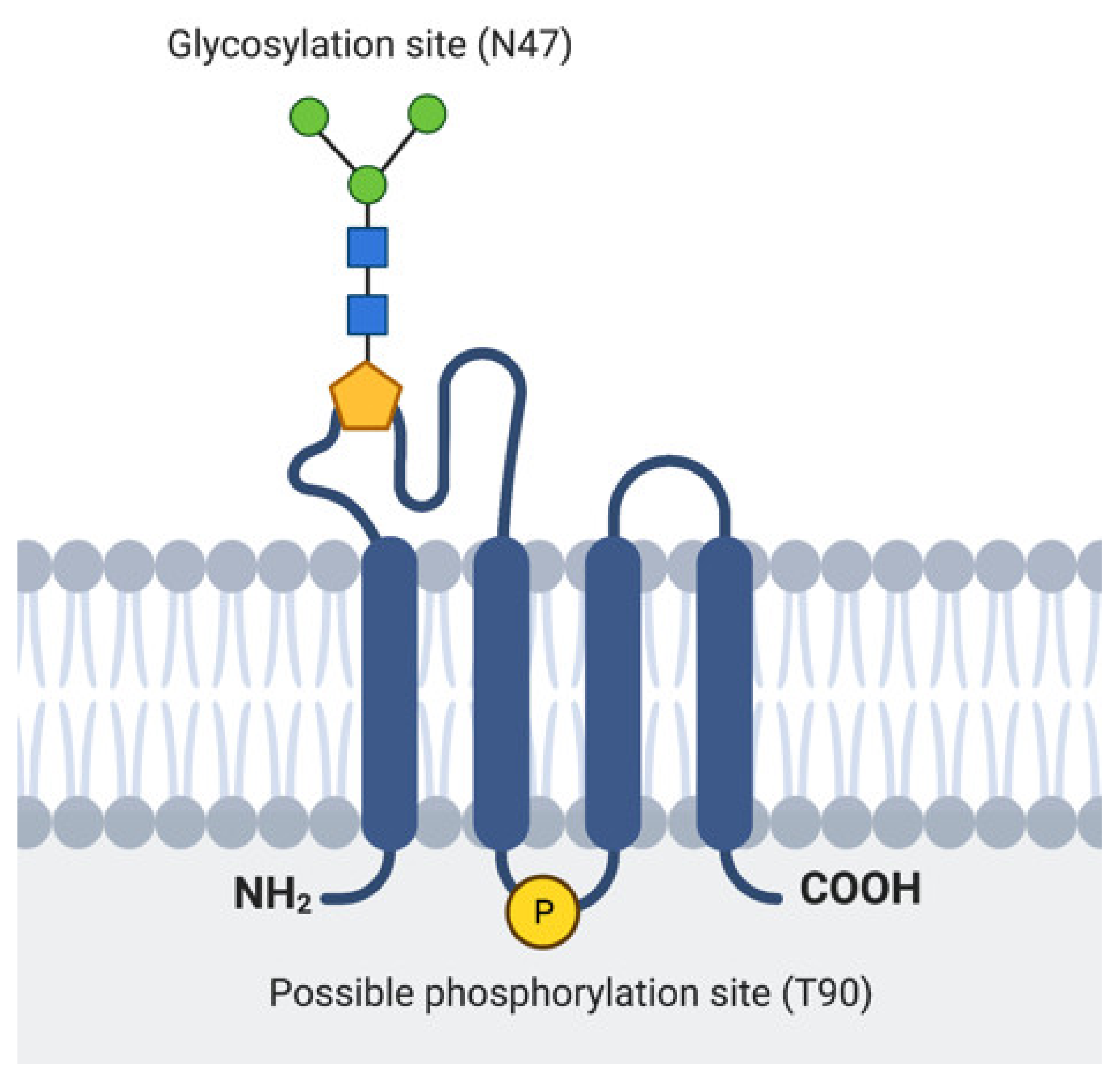
2.1. EMP3 Protein Structure and Localization
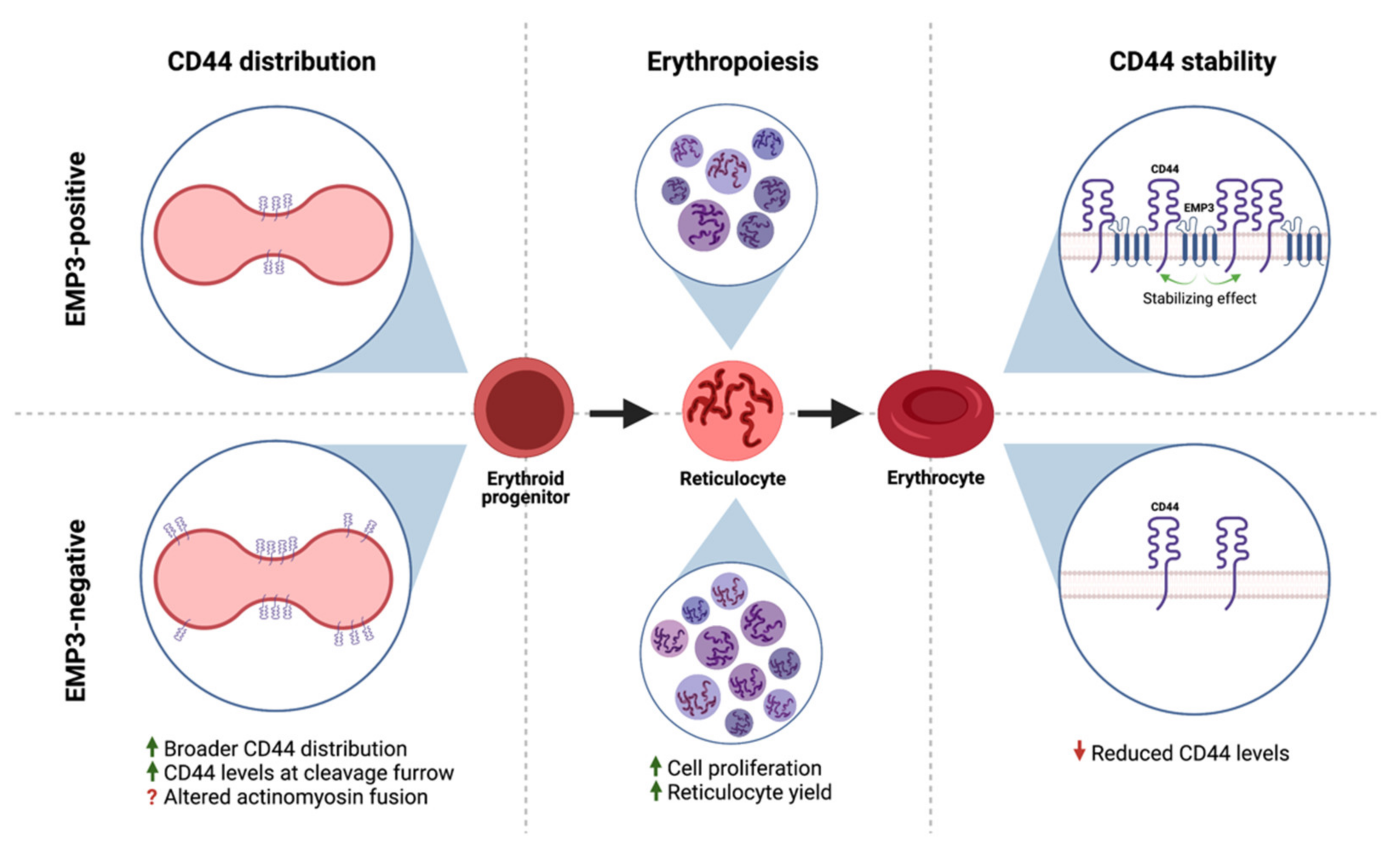
2.2. Cell Type- and Tissue-Specific Distribution of EMP3
3. EMP3 in IDH-wt GBM
3.1. EMP3 Expression in IDH-wt GBM
3.2. Prognostic Value of EMP3 in IDH-wt GBM
4. EMP3-Dependent Regulation of Membrane Receptors
4.1. Role of EMP3 in Receptor Tyrosine Kinase Signaling
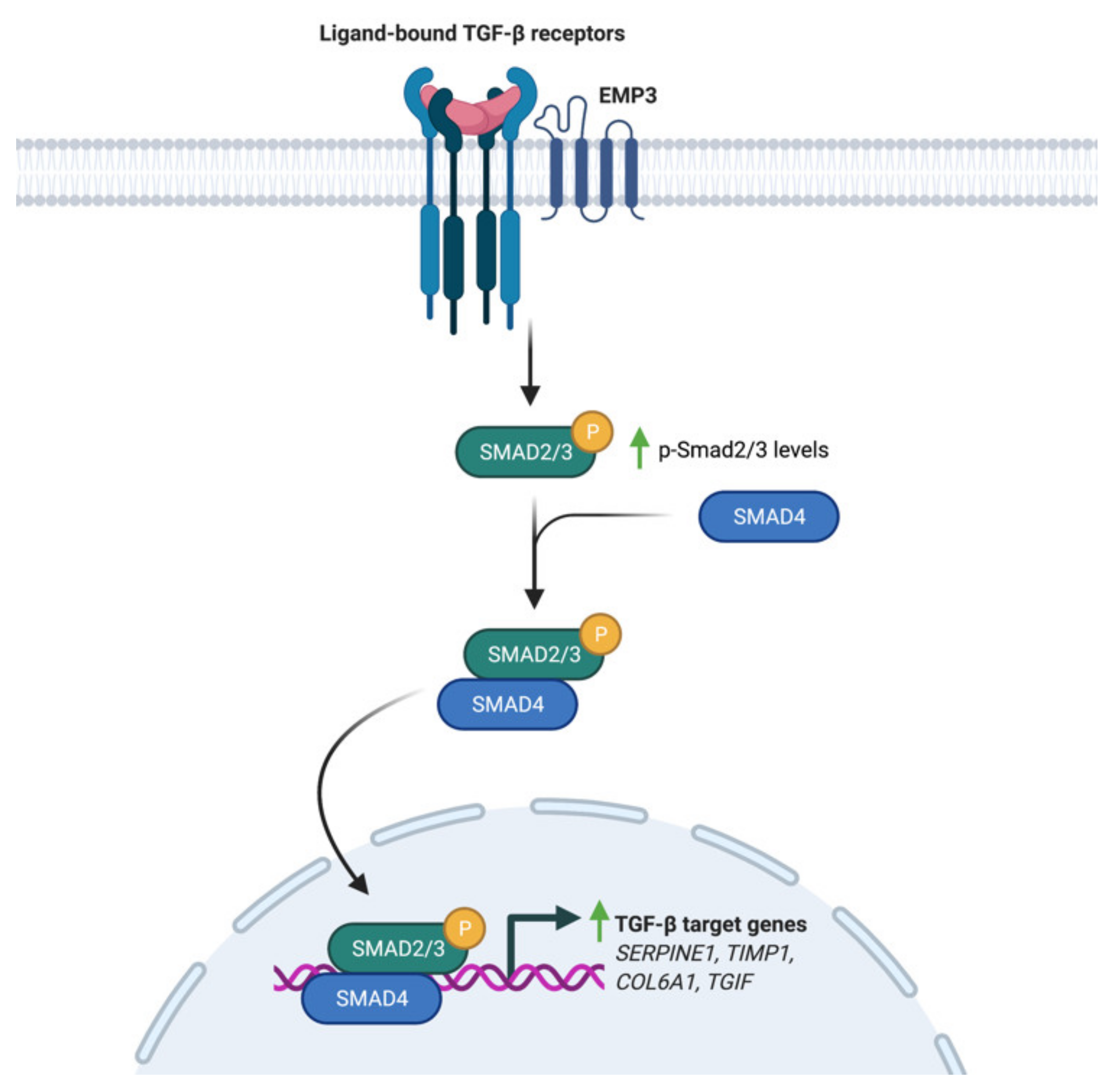
4.2. EMP3-Dependent Regulation of TGF-β Signaling
4.3. EMP3 and GBM-Associated Extracellular Matrix Receptors
4.4. Potential Involvement of the EMP3-P2RX7 Axis in GBM
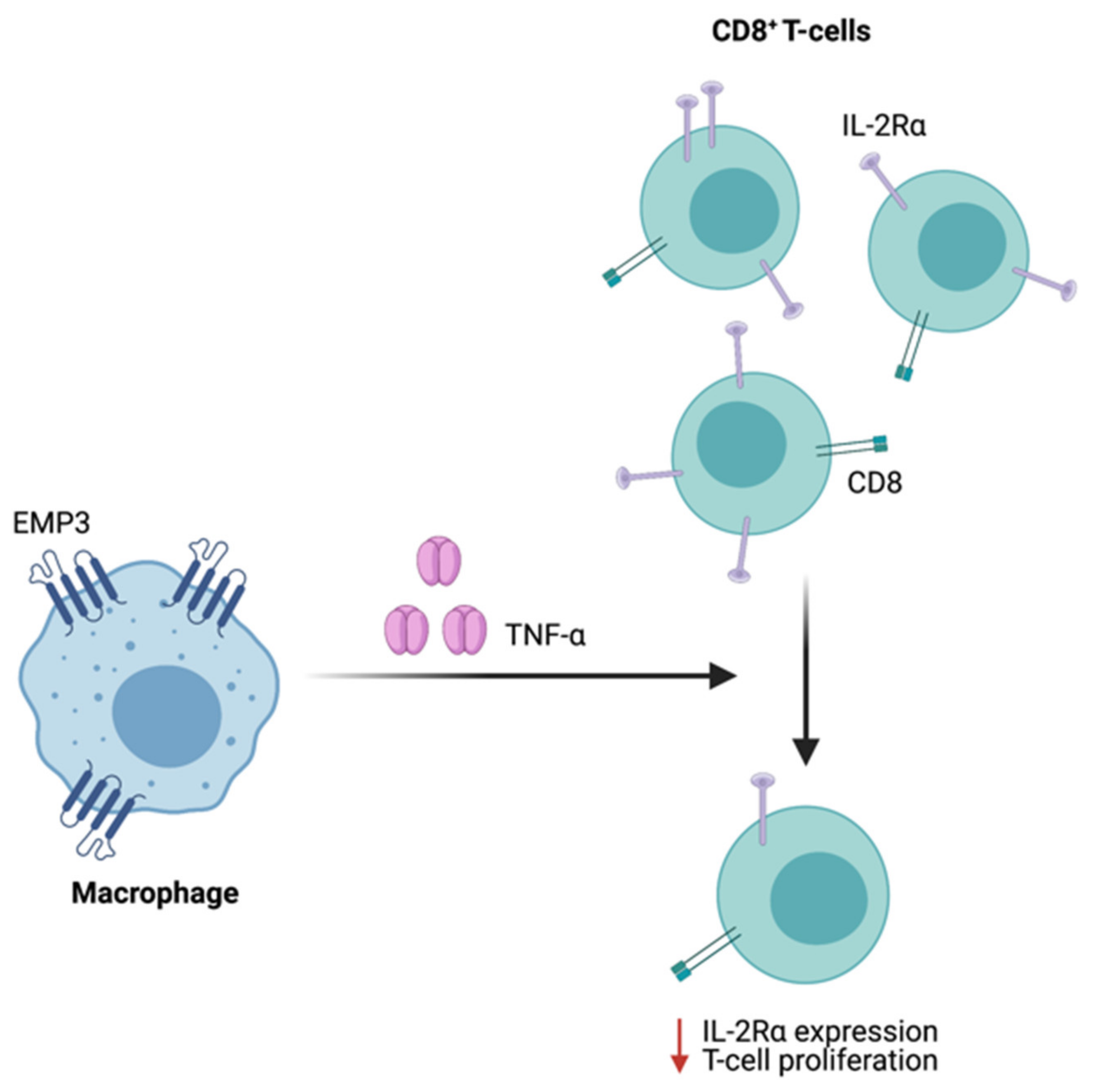
4.5. Emerging Insights on the Immunological Role of EMP3
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Rev. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Louis, D.N.; Rozenblatt-Rosen, O.; Suvà, M.L.; Regev, A.; Bernstein, B.E.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Wu, Y.; Fletcher, M.; Gu, Z.; Wang, Q.; Costa, B.; Bertoni, A.; Man, K.-H.; Schlotter, M.; Felsberg, J.; Mangei, J.; et al. Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nat. Cell Biol. 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Yue, H.; Xu, Q.; Xie, S. High EMP3 expression might independently predict poor overall survival in glioblastoma and its expression is related to DNA methylation. Medicine 2018, 97, e9538. [Google Scholar] [CrossRef]
- Jun, F.; Hong, J.; Liu, Q.; Guo, Y.; Liao, Y.; Huang, J.; Wen, S.; Shen, L. Epithelial membrane protein 3 regulates TGF-β signaling activation in CD44-high glioblastoma. Oncotarget 2016, 8, 14343–14358. [Google Scholar] [CrossRef]
- Scrideli, C.A.; Carlotti, C.G.C.; Marie, S.K.N.; Tone, L.G.; Carlotti, C.G.; Okamoto, O.K.; Andrade, V.S.; Cortez, M.A.A.; Motta, F.J.N.; Lucio-Eterovic, A.K.; et al. Gene expression profile analysis of primary glioblastomas and non-neoplastic brain tissue: Identification of potential target genes by oligonucleotide microarray and real-time quantitative PCR. J. Neuro-Oncol. 2008, 88, 281–291. [Google Scholar] [CrossRef]
- Ernst, A.; Hofmann, S.; Ahmadi, R.; Becker, N.; Korshunov, A.; Engel, F.; Hartmann, C.; Felsberg, J.; Sabel, M.; Peterziel, H.; et al. Genomic and Expression Profiling of Glioblastoma Stem Cell-Like Spheroid Cultures Identifies Novel Tumor-Relevant Genes Associated with Survival. Clin. Cancer Res. 2009, 15, 6541–6550. [Google Scholar] [CrossRef]
- Guo, X.; Su, J.; He, X. A 4-gene panel predicting the survival of patients with glioblastoma. J. Cell. Biochem. 2019, 120, 16037–16043. [Google Scholar] [CrossRef]
- Gerber, N.K.; Goenka, A.; Turcan, S.; Reyngold, M.; Makarov, V.; Kannan, K.; Beal, K.; Omuro, A.; Yamada, Y.; Gutin, P.; et al. Transcriptional diversity of long-term glioblastoma survivors. Neuro-Oncology 2014, 16, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Colman, H.; Zhang, L.; Sulman, E.P.; McDonald, J.M.; Shooshtari, N.L.; Rivera, A.; Popoff, S.; Nutt, C.L.; Louis, D.N.; Cairncross, J.G.; et al. A multigene predictor of outcome in glioblastoma. Neuro-Oncology 2009, 12, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Kozak, C.A.; Benvenisty, N. Chromosomal Mapping ofTmp(Emp1),Xmp(Emp2), andYmp(Emp3), Genes Encoding Membrane Proteins Related toPmp22. Genomics 1998, 49, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Cheng, H.-L.; Ding, Y.-R.; Chou, L.-H.; Chow, N.-H. EMP1, EMP 2, and EMP3 as novel therapeutic targets in human cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1868, 199–211. [Google Scholar] [CrossRef]
- Christians, A.; Poisel, E.; Hartmann, C.; Von Deimling, A.; Pusch, S. Characterization of the epithelial membrane protein 3 interaction network reveals a potential functional link to mitogenic signal transduction regulation. Int. J. Cancer 2019, 145, 461–473. [Google Scholar] [CrossRef]
- Taylor, V.; Suter, U. Epithelial membrane protein-2 and epithelial membrane protein-3: Two novel members of the peripheral myelin protein 22 gene family. Gene 1996, 175, 115–120. [Google Scholar] [CrossRef]
- Thornton, N.; Crew, V.K.; Tilley, L.; Green, C.A.; Tay, C.L.; Griffiths, R.E.; Singleton, B.K.; Spring, F.; Walser, P.; Alattar, A.G.; et al. Disruption of the tumour-associated EMP3 enhances erythroid proliferation and causes the MAM-negative phenotype. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Hebert, D.N.; Lamriben, L.; Powers, E.T.; Kelly, J.W. The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat. Chem. Biol. 2014, 10, 902–910. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Blal, H.A.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchís-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nat. Cell Biol. 2019, 574, 418–422. [Google Scholar] [CrossRef]
- Kunitz, A.; Wolter, M.; Boom, J.V.D.; Felsberg, J.; Tews, B.; Hahn, M.; Benner, A.; Sabel, M.; Lichter, P.; Reifenberger, G.; et al. DNA hypermethylation and Aberrant Expression of the EMP3 Gene at 19q13.3 in Human Gliomas. Brain Pathol. 2007, 17, 363–370. [Google Scholar] [CrossRef]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef]
- Hsieh, Y.-H.; Hsieh, S.-C.; Lee, C.-H.; Yang, S.-F.; Cheng, C.-W.; Tang, M.-J.; Lin, C.-L.; Lin, C.-L.; Chou, R.-H. Targeting EMP3 suppresses proliferation and invasion of hepatocellular carcinoma cells through inactivation of PI3K/Akt pathway. Oncotarget 2015, 6, 34859–34874. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Li, W.-M.; Wu, W.-J.; Chai, C.-Y.; Liu, H.-S.; Lai, M.-D.; Chow, N.-H. Potential Significance of EMP3 in Patients with Upper Urinary Tract Urothelial Carcinoma: Crosstalk with ErbB2-PI3K-Akt Pathway. J. Urol. 2014, 192, 242–251. [Google Scholar] [CrossRef]
- Zhou, W.; Jiang, Z.; Li, X.; Xu, F.; Liu, Y.; Wen, P.; Kong, L.; Hou, M.; Yu, J. EMP3 Overexpression in Primary Breast Carcinomas is not Associated with Epigenetic Aberrations. J. Korean Med. Sci. 2009, 24, 97–103. [Google Scholar] [CrossRef]
- Amaddii, M.; Meister, M.; Banning, A.; Tomasovic, A.; Mooz, J.; Rajalingam, K.; Tikkanen, R. Flotillin-1/Reggie-2 Protein Plays Dual Role in Activation of Receptor-tyrosine Kinase/Mitogen-activated Protein Kinase Signaling. J. Biol. Chem. 2012, 287, 7265–7278. [Google Scholar] [CrossRef]
- Zhang, C.; Li, A.; Zhang, X.; Xiao, H. A Novel TIP30 Protein Complex Regulates EGF Receptor Signaling and Endocytic Degradation. J. Biol. Chem. 2011, 286, 9373–9381. [Google Scholar] [CrossRef]
- Kaminska, B.; Cyranowski, S. Recent Advances in Understanding Mechanisms of TGF Beta Signaling and Its Role in Glioma Pathogenesis. In Glioma Signaling; Baranska, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 179–202. ISBN 9783030306502. [Google Scholar]
- Ark, A.V.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- D’Abaco, G.M.; Kaye, A.H. Integrins: Molecular determinants of glioma invasion. J. Clin. Neurosci. 2007, 14, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Johansson, E.; Grassi, E.S.; Pantazopoulou, V.; Tong, B.; Lindgren, D.; Berg, T.J.; Pietras, E.J.; Axelson, H.; Pietras, A. CD44 Interacts with HIF-2α to Modulate the Hypoxic Phenotype of Perinecrotic and Perivascular Glioma Cells. Cell Rep. 2017, 20, 1641–1653. [Google Scholar] [CrossRef]
- Bourguignon, L.Y. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 2008, 18, 251–259. [Google Scholar] [CrossRef]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 Signaling in the Glioma Perivascular Niche Enhances Cancer Stem Cell Phenotypes and Promotes Aggressive Tumor Growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef]
- Kan, L.K.; Williams, D.; Drummond, K.; O’Brien, T.; Monif, M. The role of microglia and P2X7 receptors in gliomas. J. Neuroimmunol. 2019, 332, 138–146. [Google Scholar] [CrossRef]
- McLarnon, J.G. Roles of purinergic P2X 7 receptor in glioma and microglia in brain tumors. Cancer Lett. 2017, 402, 93–99. [Google Scholar] [CrossRef]
- Ji, Z.; Xie, Y.; Guan, Y.; Zhang, Y.; Cho, K.-S.; Ji, M.; You, Y. Involvement of P2X7 Receptor in Proliferation and Migration of Human Glioma Cells. BioMed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Ryu, J.K.; Jantaratnotai, N.; Serrano-Perez, M.C.; McGeer, P.L.; McLarnon, J.G. Block of Purinergic P2X7R Inhibits Tumor Growth in a C6 Glioma Brain Tumor Animal Model. J. Neuropathol. Exp. Neurol. 2011, 70, 13–22. [Google Scholar] [CrossRef]
- Bergamin, L.S.; Capece, M.; Salaro, E.; Sarti, A.C.; Falzoni, S.; Pereira, M.S.L.; De Bastiani, M.A.; Scholl, J.N.; Battastini, A.M.O.; Di Virgilio, F. Role of the P2X7 receptor in in vitro and in vivo glioma tumor growth. Oncotarget 2019, 10, 4840–4856. [Google Scholar] [CrossRef]
- Ziberi, S.; Zuccarini, M.; Carluccio, M.; Giuliani, P.; Ricci-Vitiani, L.; Pallini, R.; Caciagli, F.; Di Iorio, P.; Ciccarelli, R. Upregulation of Epithelial-To-Mesenchymal Transition Markers and P2X7 Receptors Is Associated to Increased Invasiveness Caused by P2X7 Receptor Stimulation in Human Glioblastoma Stem Cells. Cells 2019, 9, 85. [Google Scholar] [CrossRef]
- Kan, L.K.; Seneviratne, S.; Drummond, K.J.; Williams, D.A.; O’Brien, T.J.; Monif, M. P2X7 receptor antagonism inhibits tumour growth in human high-grade gliomas. Purinergic Signal. 2020, 16, 327–336. [Google Scholar] [CrossRef]
- Wilson, H.L.; Wilson, S.A.; Surprenant, A.; North, R.A. Epithelial Membrane Proteins Induce Membrane Blebbing and Interact with the P2X7 Receptor C Terminus. J. Biol. Chem. 2002, 277, 34017–34023. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Mangani, D.; Weller, M.; Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharmacol. 2017, 130, 1–9. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma: Fig. 1. Neuro-Oncology 2015, 17, vii9–vii14. [Google Scholar] [CrossRef]
- Strepkos, D.; Markouli, M.; Klonou, A.; Piperi, C.; Papavassiliou, A.G. Insights in the immunobiology of glioblastoma. J. Mol. Med. 2019, 98, 1–10. [Google Scholar] [CrossRef]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9. [Google Scholar] [CrossRef]
- Kusumoto, Y.; Okuyama, H.; Shibata, T.; Konno, K.; Takemoto, Y.; Maekawa, D.; Kononaga, T.; Ishii, T.; Akashi-Takamura, S.; Saitoh, S.-I.; et al. Epithelial membrane protein 3 (Emp3) downregulates induction and function of cytotoxic T lymphocytes by macrophages via TNF-α production. Cell. Immunol. 2018, 324, 33–41. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Wu, T.; Yoon, H.; Xiong, Y.; Dixon-Clarke, S.E.; Nowak, R.P.; Fischer, E.S. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat. Struct. Mol. Biol. 2020, 27, 605–614. [Google Scholar] [CrossRef]
- Ding, Y.; Fei, Y.; Lu, B. Emerging New Concepts of Degrader Technologies. Trends Pharmacol. Sci. 2020, 41, 464–474. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nat. Cell Biol. 2020, 584, 291–297. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martija, A.A.; Pusch, S. The Multifunctional Role of EMP3 in the Regulation of Membrane Receptors Associated with IDH-Wild-Type Glioblastoma. Int. J. Mol. Sci. 2021, 22, 5261. https://doi.org/10.3390/ijms22105261
Martija AA, Pusch S. The Multifunctional Role of EMP3 in the Regulation of Membrane Receptors Associated with IDH-Wild-Type Glioblastoma. International Journal of Molecular Sciences. 2021; 22(10):5261. https://doi.org/10.3390/ijms22105261
Chicago/Turabian StyleMartija, Antoni Andreu, and Stefan Pusch. 2021. "The Multifunctional Role of EMP3 in the Regulation of Membrane Receptors Associated with IDH-Wild-Type Glioblastoma" International Journal of Molecular Sciences 22, no. 10: 5261. https://doi.org/10.3390/ijms22105261
APA StyleMartija, A. A., & Pusch, S. (2021). The Multifunctional Role of EMP3 in the Regulation of Membrane Receptors Associated with IDH-Wild-Type Glioblastoma. International Journal of Molecular Sciences, 22(10), 5261. https://doi.org/10.3390/ijms22105261