
Rare Gain-of-Function KCND3 Variant Associated with Cerebellar Ataxia, Parkinsonism, Cognitive Dysfunction, and Brain Iron Accumulation

 , , and
, , and
Abstract
1. Introduction
2. Results
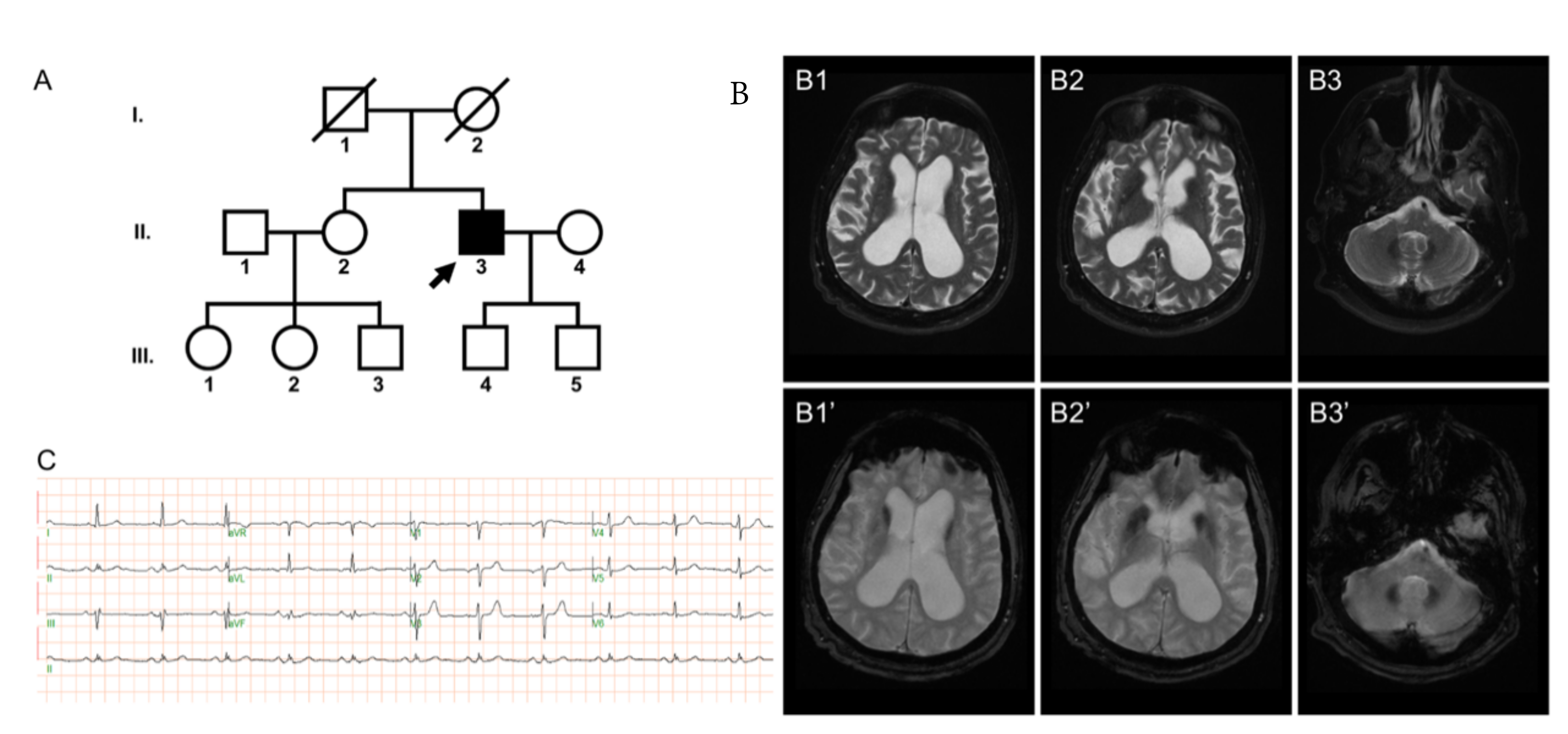
2.1. Case Presentation
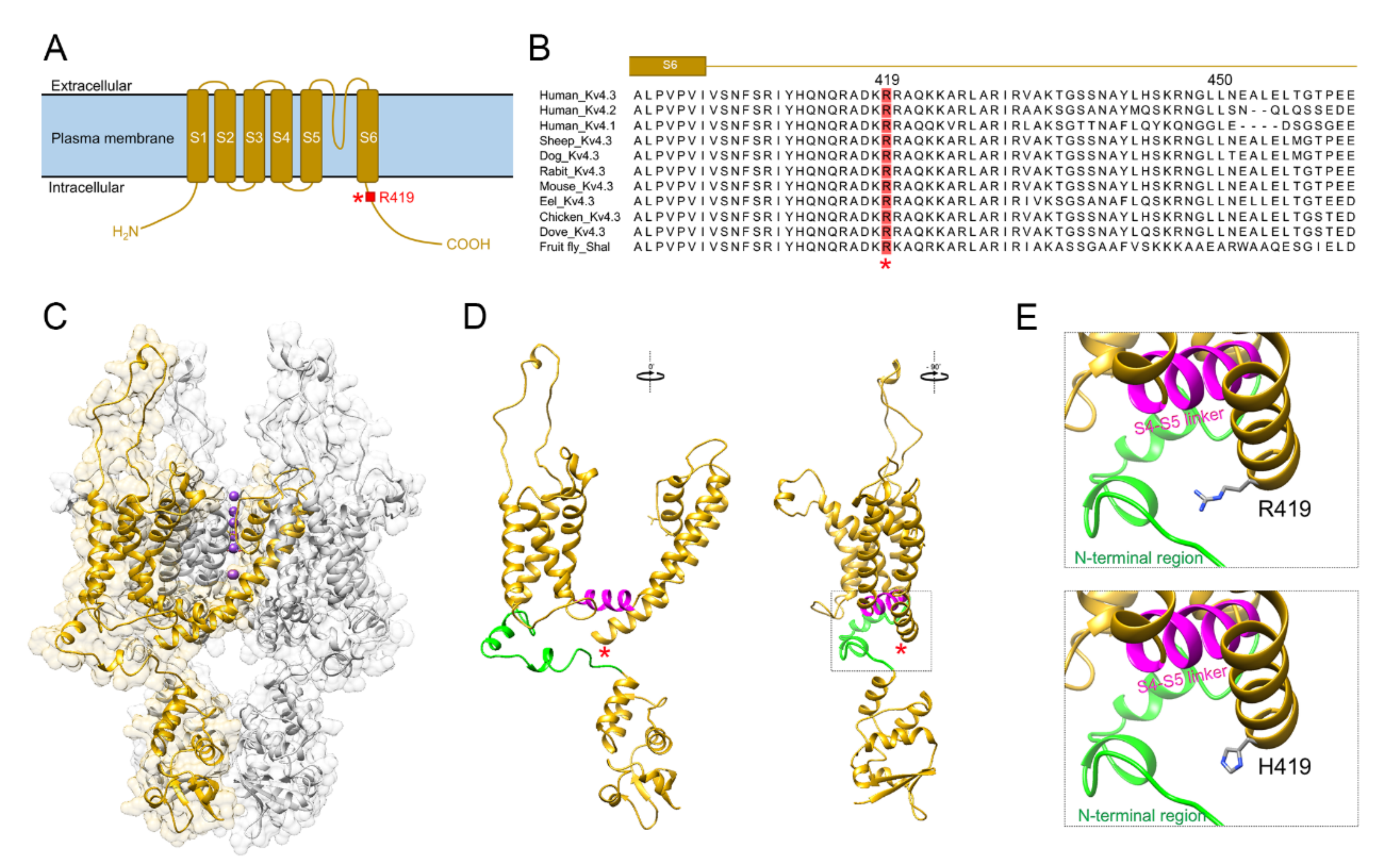
2.2. In Silico Pathogenicity
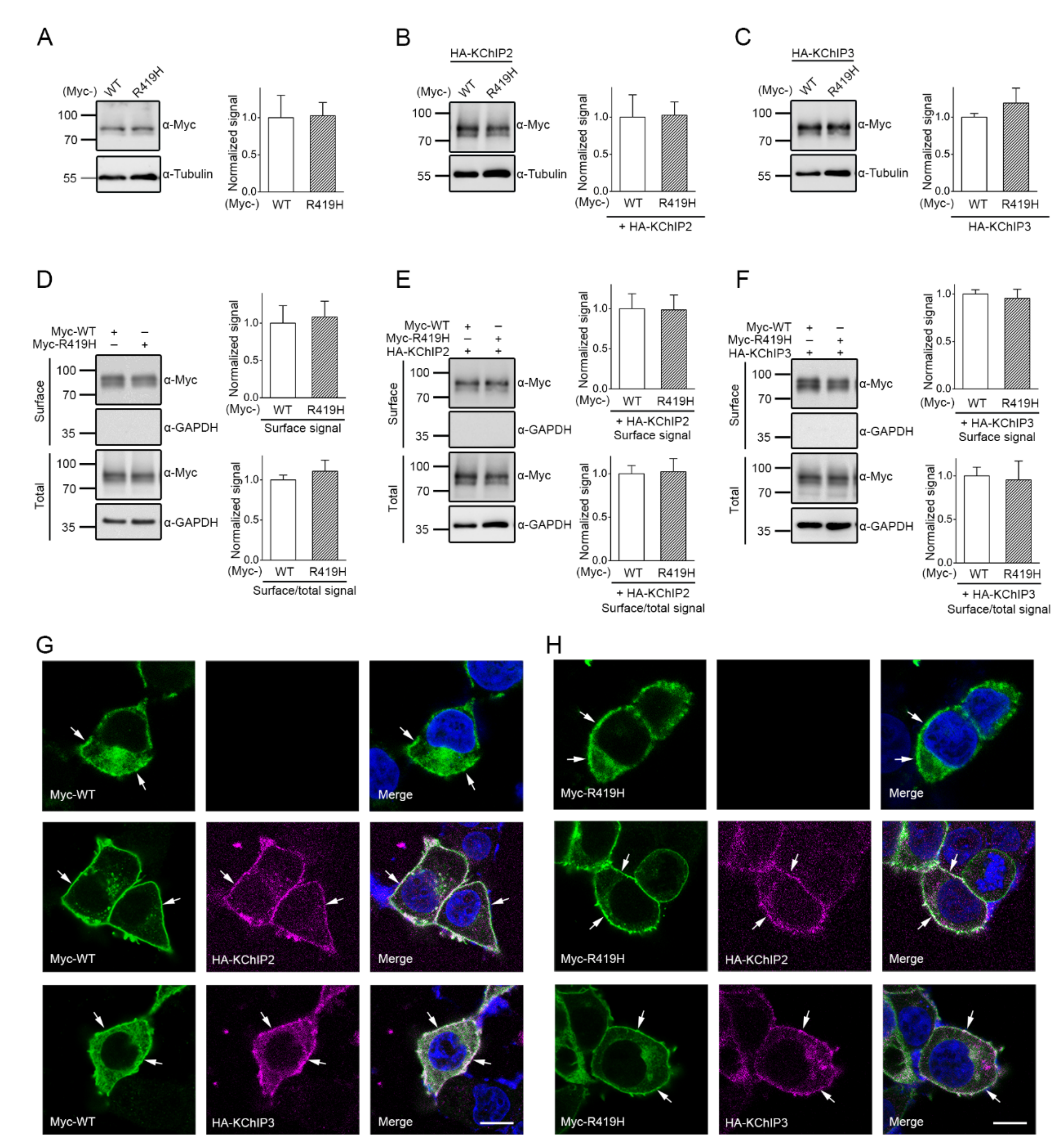
2.3. Lack of Effect of the p.R419H Variant on KV4.3 Protein Expression and Localization
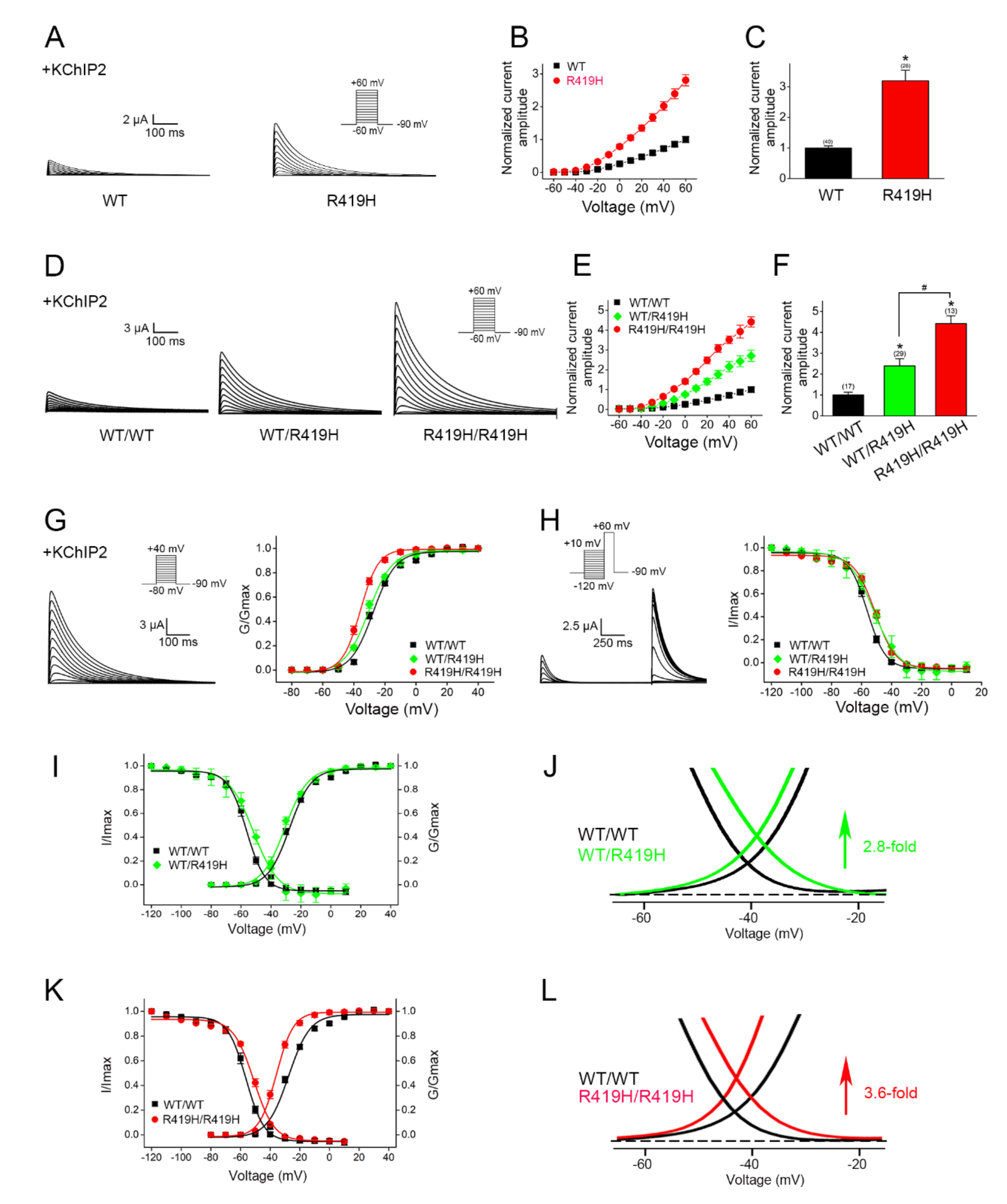
2.4. Dominant Gain-of-Function Effect of the p.R419H Variant on KV4.3 Channel Function
3. Discussion
4. Materials and Methods
4.1. Patient Evaluations and Ethics
4.2. Genetic Analyses
4.3. Bioinformatics Tools
4.4. cDNA Constructs
4.5. Cell Culture and Transfection
4.6. Immunoblotting
4.7. Immunofluorescence
4.8. Electrophysiological Analyses
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMT1 | divalent metal transporter 1 |
| ECG | electrocardiogram |
| gnomAD | genome Aggregation Database |
| HBS | HEPES buffered saline |
| HEK | human embryonic kidney |
| K+ | potassium |
| KChIP2/3 | K+ channel interacting protein 2 and 3 |
| PBS | phosphate-buffered saline |
| SCA19/22 | spinocerebellar ataxia type 19 and 22 |
| UPenn | University of Pennsylvania |
| WES | whole exome sequencing |
| WT | wild-type |
References
- Hoffman, D.A.; Magee, J.C.; Colbert, C.M.; Johnston, D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 1997, 387, 869–875. [Google Scholar] [CrossRef]
- Nadal, M.S.; Amarillo, Y.; de Miera, E.V.-S.; Rudy, B. Evidence for the presence of a novel Kv4-mediated A-type K(+) channel-modifying factor. J. Physiol. 2001, 537, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Feng, J.; Shi, H.; Pond, A.; Nerbonne, J.M.; Nattel, S. Potential Molecular Basis of Different Physiological Properties of the Transient Outward K + Current in Rabbit and Human Atrial Myocytes. Circ. Res. 1999, 84, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Le Bouter, S.; Szüts, V.; Varro, A.; Escande, D.; Nattel, S.; Demolombe, S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. 2007, 582, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Akar, F.G.; Wu, R.C.; Deschenes, I.; Armoundas, A.A.; Piacentino, V., III; Houser, S.R.; Tomaselli, G.F. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: Insights into molecular composition of ventricular Ito. Am. J. Physiology. Heart Circ. Physiol. 2004, 286, H602–H609. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Durr, A.; Majczenko, K.; Huang, Y.-H.; Liu, Y.-C.; Lien, C.-C.; Tsai, P.-C.; Ichikawa, Y.; Goto, J.; Monin, M.-L.; et al. Mutations inKCND3cause spinocerebellar ataxia type 22. Ann. Neurol. 2012, 72, 859–869. [Google Scholar] [CrossRef]
- Duarri, A.; Jezierska, J.; Fokkens, M.; Meijer, M.; Schelhaas, H.J.; den Dunnen, W.F.; van Dijk, F.; Verschuuren-Bemelmans, C.; Hageman, G.; van de Vlies, P.; et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 2012, 72, 870–880. [Google Scholar] [CrossRef]
- Duarri, A.; Lin, M.C.; Fokkens, M.R.; Meijer, M.; Smeets, C.J.; Nibbeling, E.A.; Boddeke, E.; Sinke, R.J.; Kampinga, H.H.; Papazian, D.M.; et al. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner. Cell. Mol. Life Sci. 2015, 72, 3387–3399. [Google Scholar] [CrossRef]
- Hsiao, C.T.; Fu, S.J.; Liu, Y.T.; Lu, Y.H.; Zhong, C.Y.; Tang, C.Y.; Soong, B.W.; Jeng, C.J. Novel SCA19/22-associated KCND3 mutations disrupt human K(V) 4.3 protein biosynthesis and channel gating. Hum. Mutat. 2019, 40, 2088–2107. [Google Scholar] [CrossRef] [PubMed]
- Pollini, L.; Galosi, S.; Tolve, M.; Caputi, C.; Carducci, C.; Angeloni, A.; Leuzzi, V. KCND3-Related Neurological Disorders: From Old to Emerging Clinical Phenotypes. Int. J. Mol. Sci. 2020, 21, 5802. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011, 8, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Kawabata-Iwakawa, R.; Kaneko, Y.; Hamano, S.-I.; Sano, R.; Tamura, S.; Hasegawa, H.; Kobari, T.; Kominato, Y.; Nishiyama, M.; et al. Novel Cardiocerebral Channelopathy Associated with a KCND3 V392I Mutation. Int. Heart J. 2020, 61, 1049–1055. [Google Scholar] [CrossRef]
- Takayama, K.; Ohno, S.; Ding, W.-G.; Ashihara, T.; Fukumoto, D.; Wada, Y.; Makiyama, T.; Kise, H.; Hoshiai, M.; Matsuura, H.; et al. A de novo gain-of-function KCND3 mutation in early repolarization syndrome. Heart Rhythm. 2019, 16, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Duarri, A.; Nibbeling, E.; Fokkens, M.R.; Meijer, M.; Boddeke, E.; Lagrange, E.; Stevanin, G.; Brice, A.; Durr, A.; Verbeek, D.S. The L450F [Corrected] mutation in KCND3 brings spinocerebellar ataxia and Brugada syndrome closer together. Neurogenetics 2013, 14, 257–258. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Oakley, A.E.; Collingwood, J.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, Y.; Rogers, J.T.; Wang, F. Perturbed Iron Distribution in Alzheimer’s Disease Serum, Cerebrospinal Fluid, and Selected Brain Regions: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2014, 42, 679–690. [Google Scholar] [CrossRef]
- Kruer, M.; Boddaert, N.; Schneider, S.; Houlden, H.; Bhatia, K.; Gregory, A.; Anderson, J.; Rooney, W.; Hogarth, P.; Hayflick, S. Neuroimaging Features of Neurodegeneration with Brain Iron Accumulation. Am. J. Neuroradiol. 2011, 33, 407–414. [Google Scholar] [CrossRef]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Claassen, J.; Gerding, W.M.; Kastrup, O.; Uslar, E.; Goericke, S.; Timmann, D. Excessive brain iron accumulation in spinocerebellar ataxia type 17. Neurology 2015, 84, 212–213. [Google Scholar] [CrossRef]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Moltó, M.D. The Role of Iron in Friedreich’s Ataxia: Insights From Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13, 75. [Google Scholar] [CrossRef]
- Bs, J.A.; Rogers, H.; Bs, A.H.; Snell, M.; Dill, M.; Judd, D.; Hagerman, P.; Martínez-Cerdeño, V. Iron accumulation and dysregulation in the putamen in fragile X-associated tremor/ataxia syndrome. Mov. Disord. 2017, 32, 585–591. [Google Scholar]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genom. 2013, 14, S6. [Google Scholar]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Coutelier, M.; Hammer, M.B.; Stevanin, G.; Monin, M.-L.; Davoine, C.-S.; Mochel, F.; Labauge, P.; Ewenczyk, C.; Ding, J.; Gibbs, J.R.; et al. Efficacy of Exome-Targeted Capture Sequencing to Detect Mutations in Known Cerebellar Ataxia Genes. JAMA Neurol. 2018, 75, 591–599. [Google Scholar] [CrossRef]
- Huin, V.; Strubi-Vuillaume, I.; Dujardin, K.; Brion, M.; Delliaux, M.; Dellacherie, D.; Cuvellier, J.C.; Cuisset, J.M.; Riquet, A.; Moreau, C.; et al. Expanding the phenotype of SCA19/22: Parkinsonism, cognitive impairment and epilepsy. Parkinsonism Relat. Disord. 2017, 45, 85–89. [Google Scholar] [CrossRef]
- Chung, M.-Y.; Lu, Y.-C.; Cheng, N.-C.; Soong, B.-W. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain 2003, 126, 1293–1299. [Google Scholar] [CrossRef]
- Smets, K.; Duarri, A.; Deconinck, T.; Ceulemans, B.; Van De Warrenburg, B.P.; Züchner, S.; Gonzalez, M.A.; Schüle, R.; Synofzik, M.; Van Der Aa, N.; et al. First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med. Genet. 2015, 16, 51. [Google Scholar] [CrossRef]
- Zanni, G.; Hsiao, C.T.; Fu, S.J.; Tang, C.Y.; Capuano, A.; Bosco, L.; Graziola, F.; Bellacchio, E.; Servidei, S.; Primiano, G.; et al. Novel KCND3 Variant Underlying Nonprogressive Congenital Ataxia or SCA19/22 Disrupt K(V)4.3 Protein Expression and K+ Currents with Variable Effects on Channel Properties. Int. J. Mol. Sci. 2021, 22, 4986. [Google Scholar] [CrossRef]
- Schelhaas, H.; Ippel, P.; Hageman, G.; Sinke, R.; Van Der Laan, E.; Beemer, F. Clinical and genetic analysis of a four-generation family with a distinct autosomal dominant cerebellar ataxia. J. Neurol. 2001, 248, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, J.; Xie, W.; Li, Q.; Zeng, Z.; Sui, H.; Shan, Z.; Huang, Z. A novel KCND3 mutation associated with early-onset lone atrial fibrillation. Oncotarget 2017, 8, 115503–115512. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Coarelli, G.; Monin, M.-L.; Konop, J.; Davoine, C.-S.; Tesson, C.; Valter, R.; Anheim, M.; Béhin, A.; Castelnovo, G.; et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 2017, 140, 1579–1594. [Google Scholar] [CrossRef]
- Paucar, M.; Ågren, R.; Li, T.; Lissmats, S.; Bergendal, Å.; Weinberg, J.; Nilsson, D.; Savichetva, I.; Sahlholm, K.; Nilsson, J.; et al. V374A KCND3 Pathogenic Variant Associated With Paroxysmal Ataxia Exacerbations. Neurol. Genet. 2021, 7, e546. [Google Scholar] [CrossRef]
- Paucar, M.; Bergendal, Å.; Gustavsson, P.; Nordenskjöld, M.; Laffita-Mesa, J.; Savitcheva, I.; Svenningsson, P. Novel Features and Abnormal Pattern of Cerebral Glucose Metabolism in Spinocerebellar Ataxia 19. Cerebellum 2018, 17, 465–476. [Google Scholar] [CrossRef]
- Iqbal, Z.; Rydning, S.L.; Wedding, I.M.; Koht, J.; Pihlstrøm, L.; Rengmark, A.H.; Henriksen, S.P.; Tallaksen, C.M.; Toft, M. Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. PLoS ONE 2017, 12, e0174667. [Google Scholar]
- Kurihara, M.; Ishiura, H.; Sasaki, T.; Otsuka, J.; Hayashi, T.; Terao, Y.; Matsukawa, T.; Mitsui, J.; Kaneko, J.; Nishiyama, K.; et al. Novel De Novo KCND3 Mutation in a Japanese Patient with Intellectual Disability, Cerebellar Ataxia, Myoclonus, and Dystonia. Cerebellum 2017, 17, 237–242. [Google Scholar] [CrossRef]
- Wang, Y.; Koh, K.; Namekawa, M.; Takiyama, Y. Whole-exome sequencing reveals a missense mutation in theKCND3gene in a patient with SCA19/22. Neurol. Clin. Neurosci. 2015, 3, 197–199. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ye, D.; Kritzberger, C.J.; Nesterenko, V.V.; Tester, D.J.; Antzelevitch, C.; Ackerman, M.J. Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Hum. Mutat. 2012, 33, 989–997. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Wen, Y.; Zhang, Q.; Yu, S.; Chen, Y.; Wu, X.; Zhang, Y.; Bao, X. Gene mutational analysis in a cohort of Chinese children with unexplained epilepsy: Identification of a new KCND3 phenotype and novel genes causing Dravet syndrome. Seizure 2019, 66, 26–30. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Wang, D.W.W.; Wang, Y. A Novel Gain-of-Function KCND3 Variant Associated with Brugada Syndrome. Cardiology 2020, 145, 623–632. [Google Scholar] [CrossRef]
- Choi, K.-D.; Kim, J.-S.; Kim, H.-J.; Jung, I.; Jeong, S.-H.; Lee, S.-H.; Kim, D.U.; Kim, S.-H.; Choi, S.Y.; Shin, J.-H.; et al. Genetic Variants Associated with Episodic Ataxia in Korea. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Olesen, M.S.; Refsgaard, L.; Holst, A.G.; Larsen, A.P.; Grubb, S.; Haunsø, S.; Svendsen, J.H.; Olesen, S.-P.; Schmitt, N.; Calloe, K. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc. Res. 2013, 98, 488–495. [Google Scholar] [CrossRef]
- Minassian, N.A.; Lin, M.-C.A.; Papazian, D.M. Altered Kv3.3 channel gating in early-onset spinocerebellar ataxia type 13. J. Physiol. 2012, 590, 1599–1614. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, H.; Xie, J. New Insights into the Crosstalk between NMDARs and Iron: Implications for Understanding Pathology of Neurological Diseases. Front. Mol. Neurosci. 2017, 10, 71. [Google Scholar] [CrossRef]
- Du, X.; Xu, H.; Shi, L.; Jiang, Z.; Song, N.; Jiang, H.; Xie, J. Activation of ATP-sensitive potassium channels enhances DMT1-mediated iron uptake in SK-N-SH cells in vitro. Sci. Rep. 2016, 6, 33674. [Google Scholar] [CrossRef]
- Bond, K.; Brinjikji, W.; Eckel, L.; Kallmes, D.; McDonald, R.; Carr, C. Dentate Update: Imaging Features of Entities That Affect the Dentate Nucleus. Am. J. Neuroradiol. 2017, 38, 1467–1474. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Q.; Ni, F.; Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA 2010, 107, 11352–11357. [Google Scholar] [CrossRef] [PubMed]
- Jeng, C.J.; Chen, Y.T.; Chen, Y.W.; Tang, C.Y. Dominant-negative effects of human P/Q-type Ca2+ channel mutations associated with episodic ataxia type 2. Am. J. Physiology. Cell Physiol. 2006, 290, C1209–C1220. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant at Chromosome Position: 1:112329579 C>T (GRCh37) | ||
|---|---|---|
| Substitution: c.1256G>A; p.R419H (Reference Sequence: NM_004980.4) | ||
| Tools | Results | Interpretation |
| † gnomAD | Variant found in 4/251,408 | Allele frequency: 1.591 × 10−5 |
| ‡ 1000 Genomes | Not identified | Absent in 1000 Genomes database |
| § CADD | Phred score: 28.2 | Top 0.15% most deleterious variant |
| ¶ PolyPhen-2 | Score: 1 | Probably damaging |
| †† SNPs&GO | Reliability Index: 6 | Disease-related |
| ‡‡ SIFT | Score: 0.01 | Deleterious (<0.05 deleterious) |
| §§ MutationTaster | Prob: 0.999999708078826 | Disease causing |
| (+KChIP2) | Activation | Inactivation | ||
|---|---|---|---|---|
| V0.5a (mV) | ka | V0.5i (mV) | ki | |
| WT | −27.6 | 7.2 | −56.3 | 6.1 |
| WT/p.R419H | −30.2 | 7.6 | −51.7 | 8.3 |
| p.R419H | −35.8 | 5.7 | −51.1 | 6.8 |
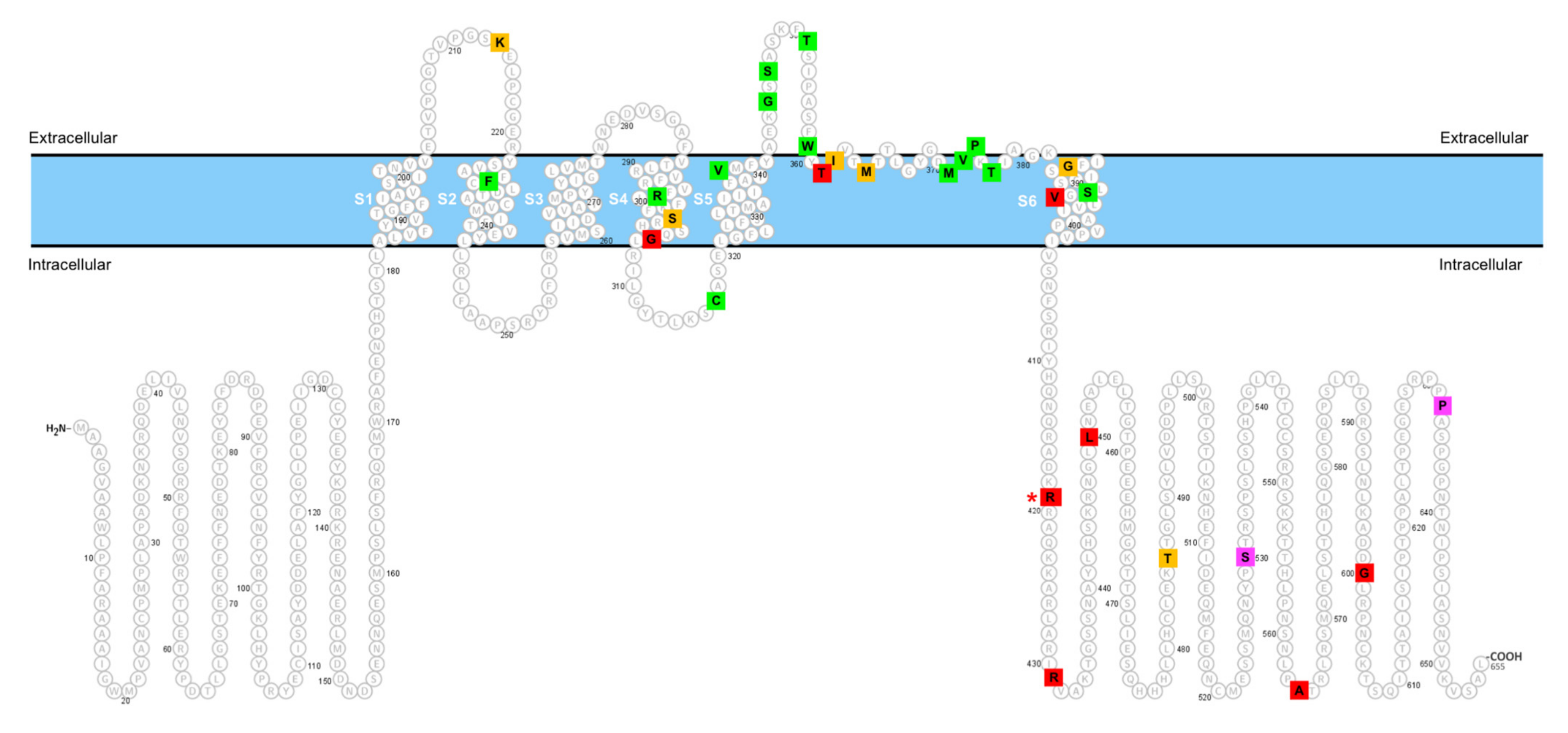
| KCND3 Variant | Clinical Feature | Remark | In Vitro Function | Reference |
|---|---|---|---|---|
| p.K214R | Episodic gait disorder, vertigo, paraesthesia, pyramidal signs, abnormal ocular movement. | An asymptomatic mother carried the variant, suggesting incomplete penetrance. | n.a. | [29] |
| p.F227 deletion | Slowly progressive cerebellar ataxia, onset from teenage to middle age; oculomotor abnormalities, pyramidal signs parkinsonism, epilepsy, or cognitive impairment have been reported in some cases. | Recurrently identified in pedigrees with autosomal dominant inheritance from multiple ethnic groups. | LOF | [6,30,31] |
| p.R293_F295 duplication | Early onset cerebellar ataxia, intellectual disability, oral apraxia, and epilepsy. | De novo mutation. | LOF | [32] |
| p.S301P | Early onset forms with neurodevelopmental disorder, epilepsy, parkinsonism-dystonia, and ataxia in adulthood | Apparently de novo mutation. | n.a. | [10] |
| p.G306A | Cardiocerebral syndrome characterized by early repolarization syndrome in combination with refractory epilepsy, and intellectual disability. | De novo mutation. | GOF | [13] |
| p.C317Y | Cerebellar ataxia onset at teenage, developmental delay, intellectual disability, myoclonus, and dystonia. | De novo mutation. | LOF | [9] |
| p.V338E | Adult-onset cerebellar ataxia; cognitive dysfunction. | Identified from an autosomal dominant inheritance pedigree. | LOF | [6,9] |
| p.G345V | Adult-onset cerebellar ataxia; variable pyramidal signs and oculomotor abnormalities. | Identified in autosomal dominant pedigrees from multiple ethnic groups. Incomplete penetrance was reported in a pedigree. | LOF | [6,33] |
| p.S347W | Adult-onset slowly progressive cerebellar ataxia. | Undetermined inheritance. | LOF | [33] |
| p.T352P | Mild cerebellar ataxia, cognitive impairment; variable degree of oculomotor disturbance, neuropathy, tremor, and myoclonus. | Identified from a large pedigree with autosomal dominant cerebellar ataxia. | LOF | [7,34] |
| p.W359G | Congenital nonprogressive ataxia; hypotonia. | De novo mutation. | LOF | [33] |
| p.T361S | Early onset lone atrial fibrillation. | One single case identified from a cohort with atrial fibrillation. | GOF | [35] |
| p.I362M | Cerebellar ataxia. | Identified from a pedigree with autosomal dominant cerebellar ataxia. | n.a. | [36] |
| p.M365T | Cerebellar ataxia. | One single case identified in an autosomal dominant cerebellar ataxia cohort study. | n.a. | [36] |
| p.M373L | Adult-onset pure cerebellar ataxia. | Two affected individuals from an autosomal dominant inheritance pedigree. | LOF | [7] |
| p.V374A | Progressive cerebellar ataxia and bradyphrenia, cognitive impairment, paroxysmal ataxia exacerbations. | Two affected individuals from an autosomal dominant inheritance pedigree. | LOF | [37] |
| p.P375S | Teenage- or adult-onset cerebellar ataxia; cognitive dysfunction, dystonia, and bradykinesia. | A symptomatic mother–son pair from an autosomal dominant inheritance pedigree. | LOF | [9] |
| p.T377M |
| A recurrently reported mutation identified in multiple ethnic groups. One single case identified from a cohort study of hereditary spastic paraplegia. | LOF | [6,9,38,39] |
| p.G384S | Cerebellar ataxia, intellectual disability, dystonia, and myoclonus. | De novo mutation. | n.a. | [40] |
| p.S390N | Teenage- or adult-onset cerebellar ataxia; cognitive dysfunction in some patients. | A recurrently reported mutation identified in multiple ethnic groups. | LOF | [7,41] |
| p.V392I |
| Identified in a case with autopsy-negative sudden unexplained death syndrome at first; one single case with Dravet syndrome was linked to the variant; a pair of siblings presented with cardiocerebral syndrome. | GOF | [12,42,43] |
| p.R419H | Slowly progressive cerebellar ataxia, parkinsonism, and cognitive dysfunction. | Identified in an apparently sporadic case. | GOF | (current study) |
| p.R431C | Episodic ataxia. | One single case from a cohort study of episodic ataxia. | n.a. | [44,45] |
| p.R431H | Brugada syndrome. | Identified from a pedigree with Brugada syndrome. | GOF | [44] |
| p.L450F |
| Identified in cases with Brugada syndrome at first; one case with autosomal dominant cerebellar ataxia was later reported. | GOF | [11,14,36] |
| p.T486A |
| One single case identified from a cohort study of hereditary spastic paraplegia. Two individuals observed in the cohort study of early-onset of persistent lone atrial fibrillation. | n.a. | [39,46] |
| p.S530P | Autopsy-negative sudden unexplained death syndrome. | Identified from a cohort with sudden unexplained death syndrome. | NSFC | [42] |
| p.A564P | Early-onset of persistent lone atrial fibrillation. | Identified from a cohort study. | GOF. | [46] |
| p.G600R (p.G581R for the short isoform) | Brugada syndrome; autopsy-negative sudden unexplained death syndrome. | Recurrently observed from patients with Brugada syndrome or sudden unexplained death syndrome. | GOF | [11,42] |
| p.P633S (p.P614S for the short isoform) | Late onset cerebellar ataxia, decreased reflexes, and vibration sense. | One single case from a cohort study of cerebellar ataxia. | NSFC | [14,36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, C.-T.; Tropea, T.F.; Fu, S.-J.; Bardakjian, T.M.; Gonzalez-Alegre, P.; Soong, B.-W.; Tang, C.-Y.; Jeng, C.-J. Rare Gain-of-Function KCND3 Variant Associated with Cerebellar Ataxia, Parkinsonism, Cognitive Dysfunction, and Brain Iron Accumulation. Int. J. Mol. Sci. 2021, 22, 8247. https://doi.org/10.3390/ijms22158247
Hsiao C-T, Tropea TF, Fu S-J, Bardakjian TM, Gonzalez-Alegre P, Soong B-W, Tang C-Y, Jeng C-J. Rare Gain-of-Function KCND3 Variant Associated with Cerebellar Ataxia, Parkinsonism, Cognitive Dysfunction, and Brain Iron Accumulation. International Journal of Molecular Sciences. 2021; 22(15):8247. https://doi.org/10.3390/ijms22158247
Chicago/Turabian StyleHsiao, Cheng-Tsung, Thomas F. Tropea, Ssu-Ju Fu, Tanya M. Bardakjian, Pedro Gonzalez-Alegre, Bing-Wen Soong, Chih-Yung Tang, and Chung-Jiuan Jeng. 2021. "Rare Gain-of-Function KCND3 Variant Associated with Cerebellar Ataxia, Parkinsonism, Cognitive Dysfunction, and Brain Iron Accumulation" International Journal of Molecular Sciences 22, no. 15: 8247. https://doi.org/10.3390/ijms22158247
APA StyleHsiao, C.-T., Tropea, T. F., Fu, S.-J., Bardakjian, T. M., Gonzalez-Alegre, P., Soong, B.-W., Tang, C.-Y., & Jeng, C.-J. (2021). Rare Gain-of-Function KCND3 Variant Associated with Cerebellar Ataxia, Parkinsonism, Cognitive Dysfunction, and Brain Iron Accumulation. International Journal of Molecular Sciences, 22(15), 8247. https://doi.org/10.3390/ijms22158247