
Identification of Pyruvate Dehydrogenase E1 as a Potential Target against Magnaporthe oryzae through Experimental and Theoretical Investigation

 and
and
Abstract
1. Introduction
2. Results
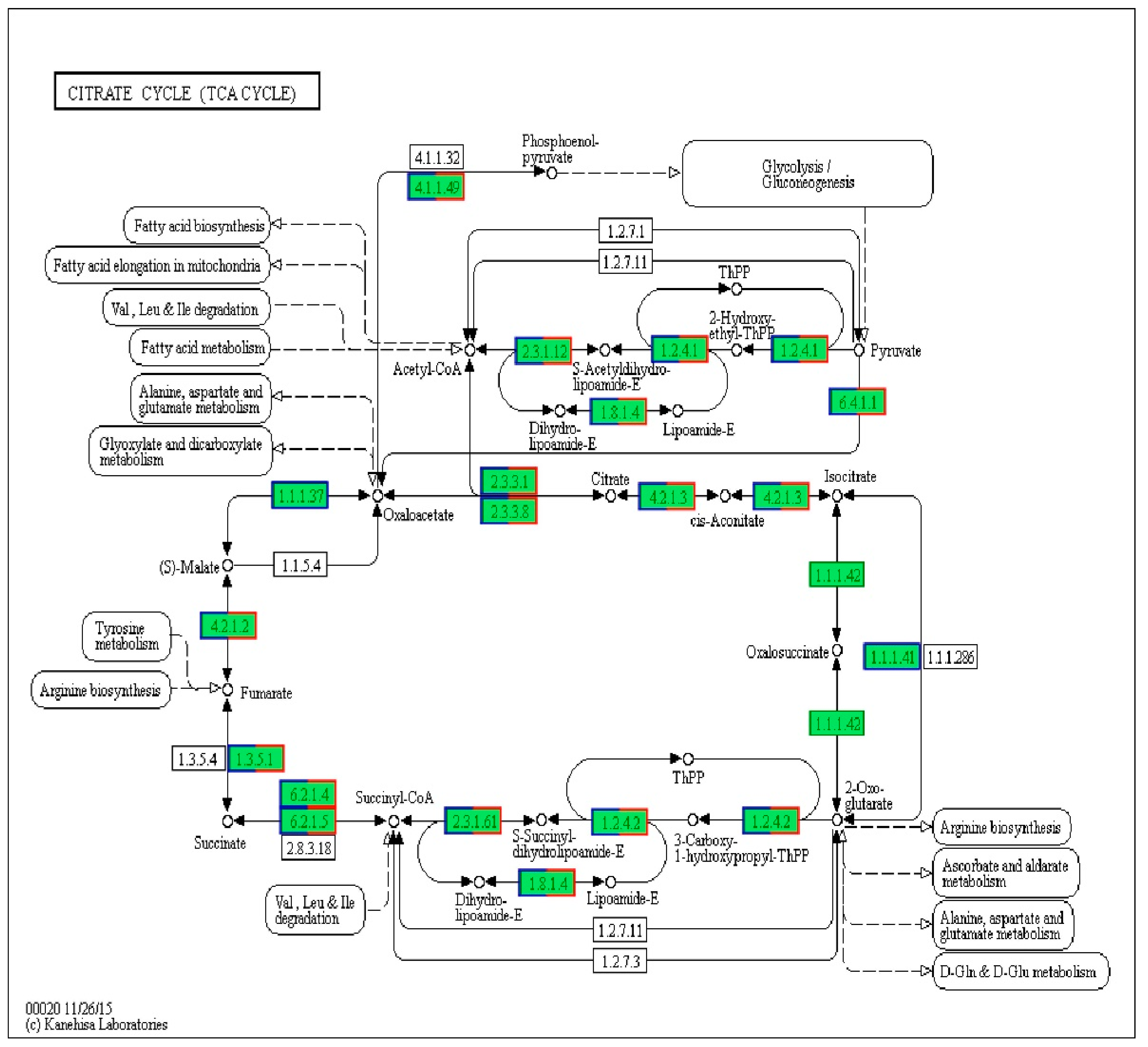
2.1. Analysis of Differentially Expressed Genes
2.2. Validation of Differentially Expressed Genes by qPCR
2.3. Enzymatic ActivityThe
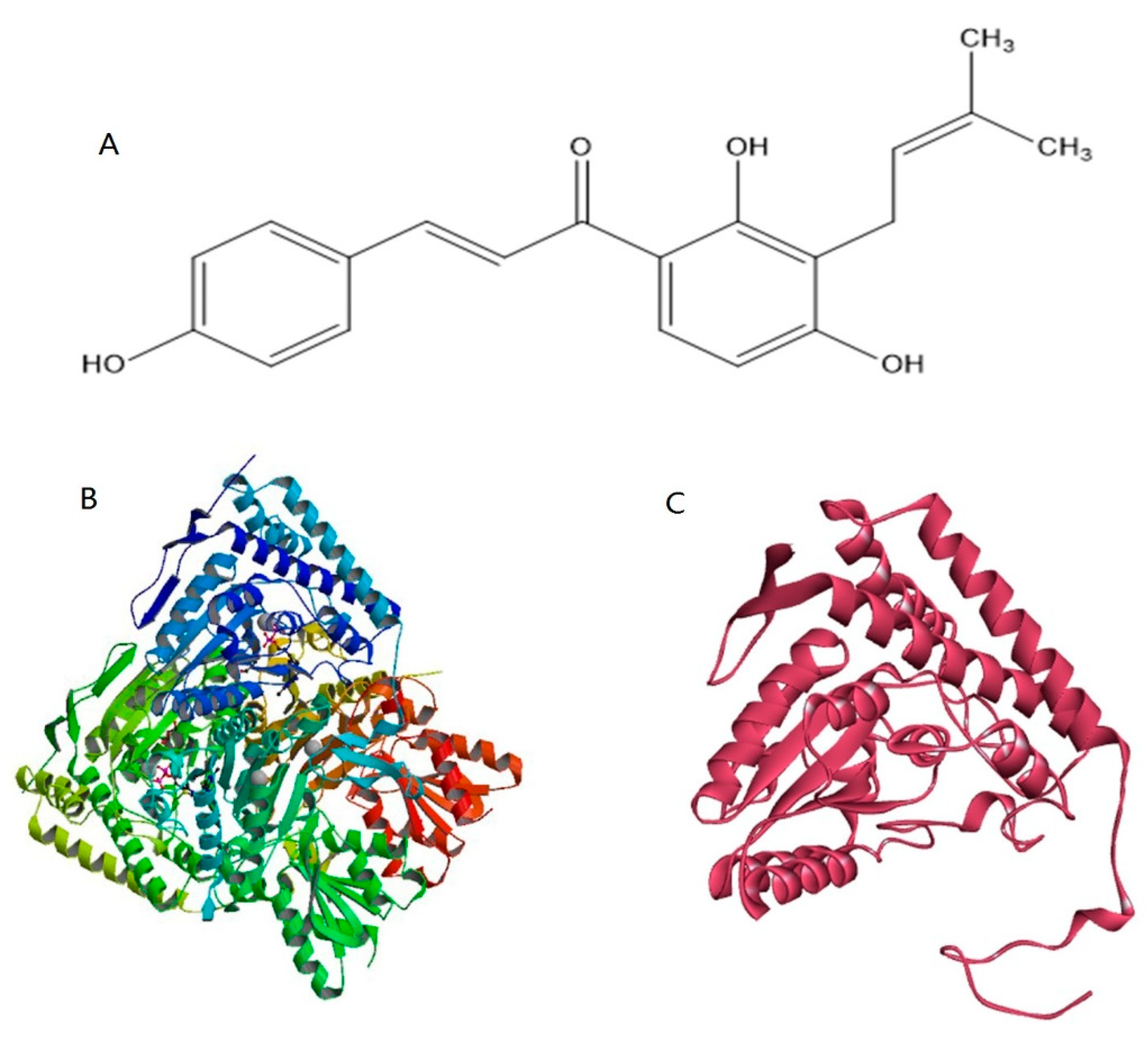
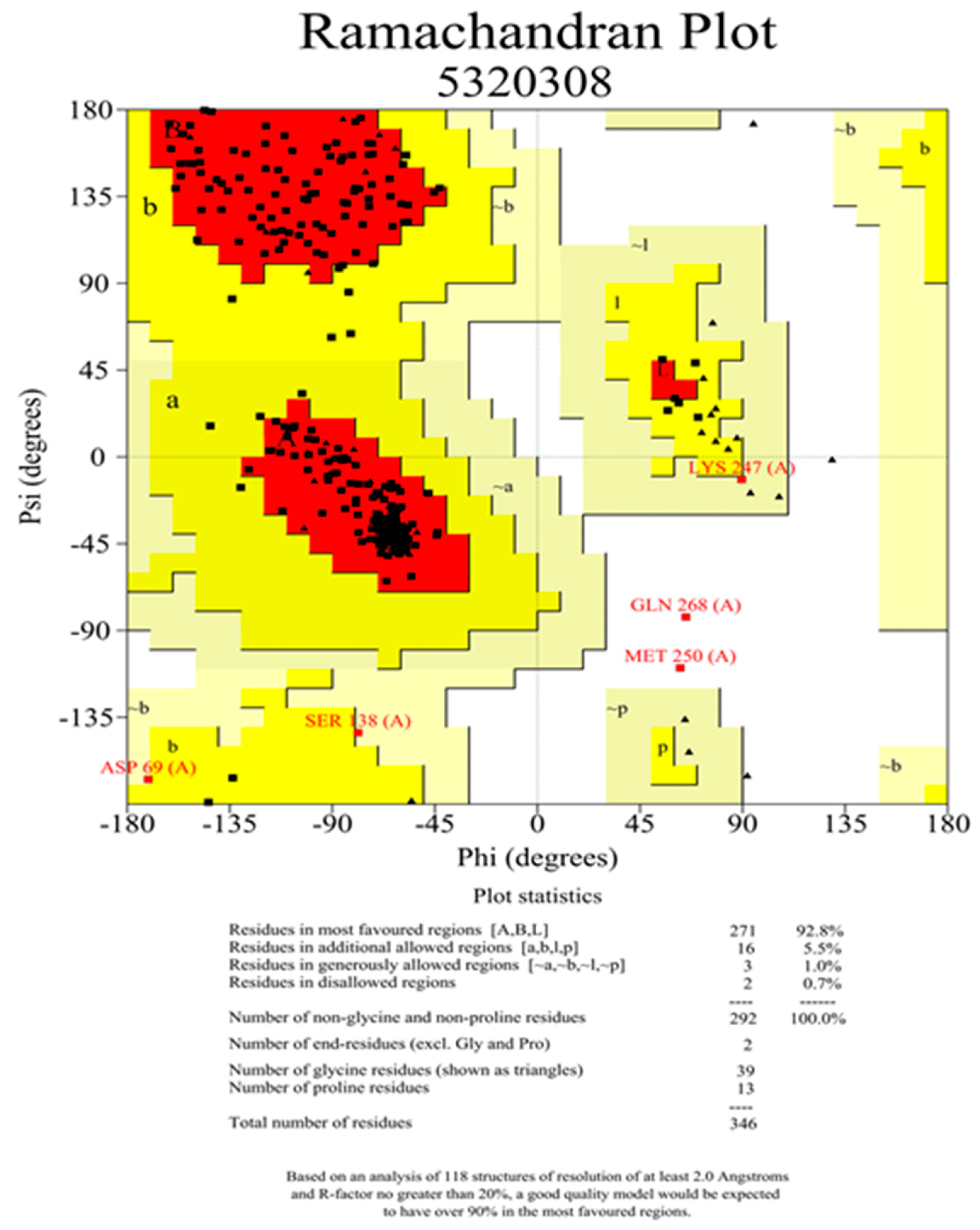
2.4. Homology Modeling and Prediction of the Active Site of Pyruvate Dehydrogenase
2.5. Protein-Ligand Docking Analysis
2.6. Molecular Dynamics Simulation of the Pyruvate Dehydrogenase E1-IBC Complexes and Binding Free Energy
3. Discussion
4. Materials and Methods
4.1. Fungal Strain
4.2. Preparation of M. oryzae conidia
4.3. Transcriptome Sequencing
4.4. Detection by Quantitative Real-Time PCR
4.5. Extraction of Solution for Enzyme Assays
4.6. Enzyme Activity Assays
4.7. Preparation of the Ligand Structure
4.8. Homology Modeling
4.9. Identification of Pyruvate Dehydrogenase Active Site
4.10. Molecular Docking Studies
4.11. Molecular Dynamics Simulations
4.12. Binding Free Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.; Chern, M.; Yin, J.; Wang, J.; Chen, X. Recent advances in broad-spectrum resistance to the rice blast disease. Curr. Opin. Plant. Biol. 2019, 50, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zou, L.; Zhu, X.; Cao, Y.; He, M.; Chen, X. Fighting the enemy: How rice survives the blast pathogen’s attack. Crop. J. 2021. [Google Scholar] [CrossRef]
- Shahriar, S.A.; Imtiaz, A.A.; Hossain, B.; Husna, A.; Eaty, M.N.K. Review: Rice Blast Disease. Ann. Res. Rev. Biol. 2020, 35, 50–64. [Google Scholar] [CrossRef]
- Yang, W.J.; Chen, J.H.; Chen, G.N.; Wang, S.H.; Fu, F.F. The early diagnosis and fast detection of blast fungus, Magnaporthe grisea, in rice plant by using its chitinase as biochemical marker and a rice cDNA encoding mannose-binding lectin as recog-nition probe. Biosens. Bioelectron. 2013, 41, 820–826. [Google Scholar] [CrossRef] [PubMed]
- He, J.-B.; He, H.-F.; Zhao, L.-L.; Zhang, L.; You, G.-Y.; Feng, L.-L.; Wan, J.; He, H.-W. Synthesis and antifungal activity of 5-iodo-1,4-disubstituted-1,2,3-triazole derivatives as pyruvate dehydrogenase complex E1 inhibitors. Bioorganic Med. Chem. 2015, 23, 1395–1401. [Google Scholar] [CrossRef]
- Dar, M.S.; Ganaie, S.A.; Raja, W.; Teeli, R.A. In-vivo investigation on antifungal properties of leaf extracts of certain medicinal plants through seed treatment and foliar sprays against rice blast disease (Magnaporthe grisea) in Kashmir, India. Ann. Agrar. Sci. 2018, 16, 267–271. [Google Scholar] [CrossRef]
- Yoon, M.-Y.; Cha, B.; Kim, J.-C. Recent Trends in Studies on Botanical Fungicides in Agriculture. Plant. Pathol. J. 2013, 29, 1–9. [Google Scholar] [CrossRef]
- Tleuova, A.B.; Wielogorska, E.; Talluri, V.P.; Štěpánek, F.; Elliott, C.T.; Grigoriev, D.O. Recent advances and remaining barriers to producing novel formulations of fungicides for safe and sustainable agriculture. J. Control. Release 2020, 326, 468–481. [Google Scholar] [CrossRef]
- Alam, F.; Khan, G.N.; Bin Asad, M.H.H. Psoralea corylifoliaL: Ethnobotanical, biological, and chemical aspects: A review. Phytother. Res. 2018, 32, 597–615. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, W.; Wang, Y.; Lu, J.; Chen, X. The Chemical Constituents and Bioactivities of Psoralea corylifolia Linn.: A Review. Am. J. Chin. Med. 2016, 44, 35–60. [Google Scholar] [CrossRef]
- Zhu, G.; Luo, Y.; Xu, X.; Zhang, H.; Zhu, M. Anti-diabetic compounds from the seeds of Psoralea corylifolia. Fitotherapy 2019, 139, 104373. [Google Scholar] [CrossRef]
- Latha, P.; Evans, D.; Panikkar, K.; Jayavardhanan, K. Immunomodulatory and antitumour properties of Psoralea corylifolia seeds. Fitotherapy 2000, 71, 223–231. [Google Scholar] [CrossRef]
- Seo, E.; Oh, Y.S.; Kim, N.; Lee, M.-Y.; Chae, S.; Jun, H.-S. Protective Role ofPsoralea corylifoliaL. Seed Extract against Hepatic Mitochondrial Dysfunction Induced by Oxidative Stress or Aging. Evid. Based Complement. Altern. Med. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Hu, B.; Wang, M.; Wang, J.; Guan, L. Identification of isobavachalcone as a potential drug for rice blast disease caused by the fungus Magnaporthe grisea. J. Biomol. Struct. Dyn. 2018, 37, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant. Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Mochida, S.; Tomimoto, D.; Konuma, T.; Kiyota, N.; Tsuda, S.; Shiga, Y.; Omodaka, K.; Nakazawa, T.A. pyruvate dehydrogenase kinase inhibitor prevents retinal cell death and improves energy metabolism in rat retinas after ische-mia/reperfusion injury. Exp. Eye Res. 2020, 193, 107997. [Google Scholar] [CrossRef] [PubMed]
- Ebertowska, A.; Ludkiewicz, B.; Klejbor, I.; Melka, N.; Morys, J. Pyruvate dehydrogenase deficiency-morphological and metabolic effects, creation of animal model to study and research for treatment therapy. Folia Morphol 2020, 79, 191–197. [Google Scholar] [CrossRef]
- He, H.-W.; Peng, H.; Wang, T.; Wang, C.; Yuan, J.-L.; Chen, T.; He, J.; Tan, X. α-(Substituted-phenoxyacetoxy)-α-heterocyclylmethylphosphonates: Synthesis, Herbicidal Activity, Inhibition on Pyruvate Dehydrogenase Complex (PDHc), and Application as Postemergent Herbicide against Broadleaf Weeds. J. Agric. Food Chem. 2013, 61, 2479–2488. [Google Scholar] [CrossRef]
- He, H.-W.; Yuan, J.-L.; Peng, H.; Chen, T.; Shen, P.; Wan, S.-Q.; Li, Y.; Tan, H.-L.; He, Y.-H.; He, J.-B.; et al. Studies of O,O-Dimethyl α-(2,4-Dichlorophenoxyacetoxy)ethylphosphonate (HW02) as a New Herbicide. 1. Synthesis and Herbicidal Activity of HW02 and Analogues as Novel Inhibitors of Pyruvate Dehydrogenase Complex. J. Agric. Food Chem. 2011, 59, 4801–4813. [Google Scholar] [CrossRef]
- Chiu, C.C.; Jordan, F. Novel Synthesis of 2-Oxo-4-phenyl-3-butynoic Acid, a New Inhibitor and Alternate Substrate of Pyruvate Decarboxylase. J. Org. Chem. 1994, 59, 5763–5766. [Google Scholar] [CrossRef]
- Pagliaccia, D.; Urak, R.Z.; Wong, F.; Douhan, L.A.I.; Greer, C.A.; Vidalakis, G.; Douhan, G.W. Genetic Structure of the Rice Blast Pathogen (Mag-naporthe oryzae) over a Decade in North Central California Rice Fields. Microb. Ecol. 2017, 75, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Rossolini, G.M.; Thaller, M.C. Coping with antibiotic resistance: Contributions from genomics. Genome Med. 2010, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; He, H.; Zhou, Y.; Cai, M.; Peng, H.; Liu, H.; Liu, L.; Feng, L.; He, H. Structure optimization and bioactivity evaluation of ThDP analogs targeting cyanobacterial pyruvate dehydrogenase E1. Bioorganic Med. Chem. 2019, 27, 115159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, J.; Zhu, Y.; Ni, S.; Chen, J.; Feng, X.; Zhang, Y.; Li, S.; Zhu, H.; Wen, Y. De novo assembly of the Camellia ni-tidissima transcriptome reveals key genes of flower pigment biosynthesis. Front. Plant Sci. 2017, 8, 1545. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.; Yan, Y.; Zhang, Z.; Brown, A.M.; Arjunan, P.; Furey, W.; Guest, J.R.; Jordan, F. Inhabition of the Escherichia coli Pyruvate Dehydrogenase Complex E1 Subunit and Its Tyrosine 177 Variants by Thiamin 2-Thiazolone and Thiamin 2-Thiothiazolone Diphosphates. J. Biol. Chem. 2001, 276, 45969–45978. [Google Scholar] [CrossRef] [PubMed]
- Tripatara, A.; Korotchkina, L.G.; Patel, M.S. Characterization of Point Mutations in Patients with Pyruvate Dehydrogenase Deficiency: Role of Methionine-181, Proline-188, and Arginine-349 in the α Subunit. Arch. Biochem. Biophys. 1999, 367, 39–50. [Google Scholar] [CrossRef]
- Alvarez, L.D.; Pecci., A. Structure and dynamics of neurosteroid binding to the α1β2γ2 GABAA receptor. J. Steroid. Biochem. Mol. Biol. 2018, 182, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Suprianto; Budiarsa, M.; Dhafir, F. 3D Structure of VP1 Structural Protein on Enterovirus A71 Using Swiss-Model. BI-OEDUSCIENCE. J. Pendidik. Biol. Sains 2020, 4, 37–47. [Google Scholar]
- Piplani, S.; Saini, V.; Niraj, R.R.K.; Pushp, A.; Kumar, A. Homology modelling and molecular docking studies of human pla-cental cadherin protein for its role in teratogenic effects of anti-epileptic drugs. Comput. Biol. Chem. 2016, 60, 1–8. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2010, 30, S162–S173. [Google Scholar] [CrossRef]
- Subhani, S.; Jayaraman, A.; Jamil, K. Homology modelling and molecular docking of MDR1 with chemotherapeutic agents in non-small cell lung cancer. Biomed. Pharmacother. 2015, 71, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Abrigach, F.; Rokni, Y.; Takfaoui, A.; Khoutoul, M.; Doucet, H.; Asehraou, A.; Touzani, R. In vitro screening, homology modeling and molecular docking studies of some pyrazole and imidazole derivatives. Biomed. Pharmacother. 2018, 103, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking. Methods Mol. Biol. 2008, 443, 365–382. [Google Scholar] [CrossRef]
- Şöhretoğlu, D.; Sari, S.; Šoral, M.; Barut, B.; Özel, A.; Liptaj, T. Potential of Potentilla inclinata and its polyphenolic compounds in α-glucosidase inhibition: Kinetics and interaction mechanism merged with docking simulations. Int. J. Biol. Macromol. 2018, 108, 81–87. [Google Scholar] [CrossRef]
- Wang, Q.; He, J.; Wu, D.; Wang, J.; Yan, J.; Li, H. Interaction of α-cyperone with human serum albumin: Determination of the binding site by using Discovery Studio and via spectroscopic methods. J. Lumin. 2015, 164, 81–85. [Google Scholar] [CrossRef]
- Puranik, N.; Srivastava, P.; Swami, S.; Choudhair, A.; Sarkar, D. Molecular modeling studies and in vitro screening of dihy-drorugosaflavonoid and its derivatives against Mycobacterium tuberculosis. RSC Adv. 2018, 8, 10634–10643. [Google Scholar] [CrossRef]
- Sheikh, I.A.; Jiffri, E.H.; Ashraf, G.M.; Kamal, M.A. Structural insights into the camel milk lactoperoxidase: Homology modeling and molecular dynamics simulation studies. J. Mol. Graph. Model. 2019, 86, 43–51. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Definition | FC (C/B) | |Log2 (FC)| |
|---|---|---|---|
| 4.1.1.49 | Phosphate carboxykinase (ATP) | 0.126 | 2.989 |
| 1.2.4.1 | Pyruvate dehydrogenase E1 component | 0.139 | 2.847 |
| 2.3.2.12 | Pyruvate dehydrogenase E2 component (dihydrolipoamide acetyltransferase) | 0.189 | 2.404 |
| 1.8.1.4 | Dihydrolipoamide dehydrogenase | 0.126 | 2.989 |
| 6.4.1.1 | Pyruvate carboxylase | 0.069 | 3.857 |
| 2.3.3.1 | Citrate synthase | 0.071 | 3.816 |
| 2.3.3.8 | ATP citrate (pro-S)-lyase | 0.086 | 3.540 |
| 4.2.1.3 | Aconitate hydratase | 0.187 | 2.419 |
| 1.1.1.41 | Isocitrate dehydrogenase (NAD+) | - | - |
| 1.1.1.42 | Isocitrate dehydrogenase | - | - |
| 1.2.4.2 | 2-Oxoglutarate dehydrogenase E1 component | 0.105 | 3.252 |
| 2.3.1.61 | 2-Oxoglutarate dehydrogenase E2 component (dihydrolipoamide succinyltransferase) | 0.154 | 2.699 |
| 4.2.1.2 | Fumarate hydratase, class I | 0.144 | 2.786 |
| 1.3.5.1 | Succinate dehydrogenase (ubiquinone) flavoprotein subunit | 0.149 | 2.796 |
| 6.2.1.4 | Succinyl-CoA synthetase alpha subunit | 0.119 | 3.071 |
| 6.2.1.5 | Succinyl-CoA synthetase alpha subunit | 0.119 | 3.071 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hu, B.; Wang, Z.; He, J.; Zhang, Y.; Wang, J.; Guan, L. Identification of Pyruvate Dehydrogenase E1 as a Potential Target against Magnaporthe oryzae through Experimental and Theoretical Investigation. Int. J. Mol. Sci. 2021, 22, 5163. https://doi.org/10.3390/ijms22105163
Li Y, Hu B, Wang Z, He J, Zhang Y, Wang J, Guan L. Identification of Pyruvate Dehydrogenase E1 as a Potential Target against Magnaporthe oryzae through Experimental and Theoretical Investigation. International Journal of Molecular Sciences. 2021; 22(10):5163. https://doi.org/10.3390/ijms22105163
Chicago/Turabian StyleLi, Yuejuan, Baichun Hu, Zhibin Wang, Jianhua He, Yaoliang Zhang, Jian Wang, and Lijie Guan. 2021. "Identification of Pyruvate Dehydrogenase E1 as a Potential Target against Magnaporthe oryzae through Experimental and Theoretical Investigation" International Journal of Molecular Sciences 22, no. 10: 5163. https://doi.org/10.3390/ijms22105163
APA StyleLi, Y., Hu, B., Wang, Z., He, J., Zhang, Y., Wang, J., & Guan, L. (2021). Identification of Pyruvate Dehydrogenase E1 as a Potential Target against Magnaporthe oryzae through Experimental and Theoretical Investigation. International Journal of Molecular Sciences, 22(10), 5163. https://doi.org/10.3390/ijms22105163