
Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development


Abstract
1. Introduction
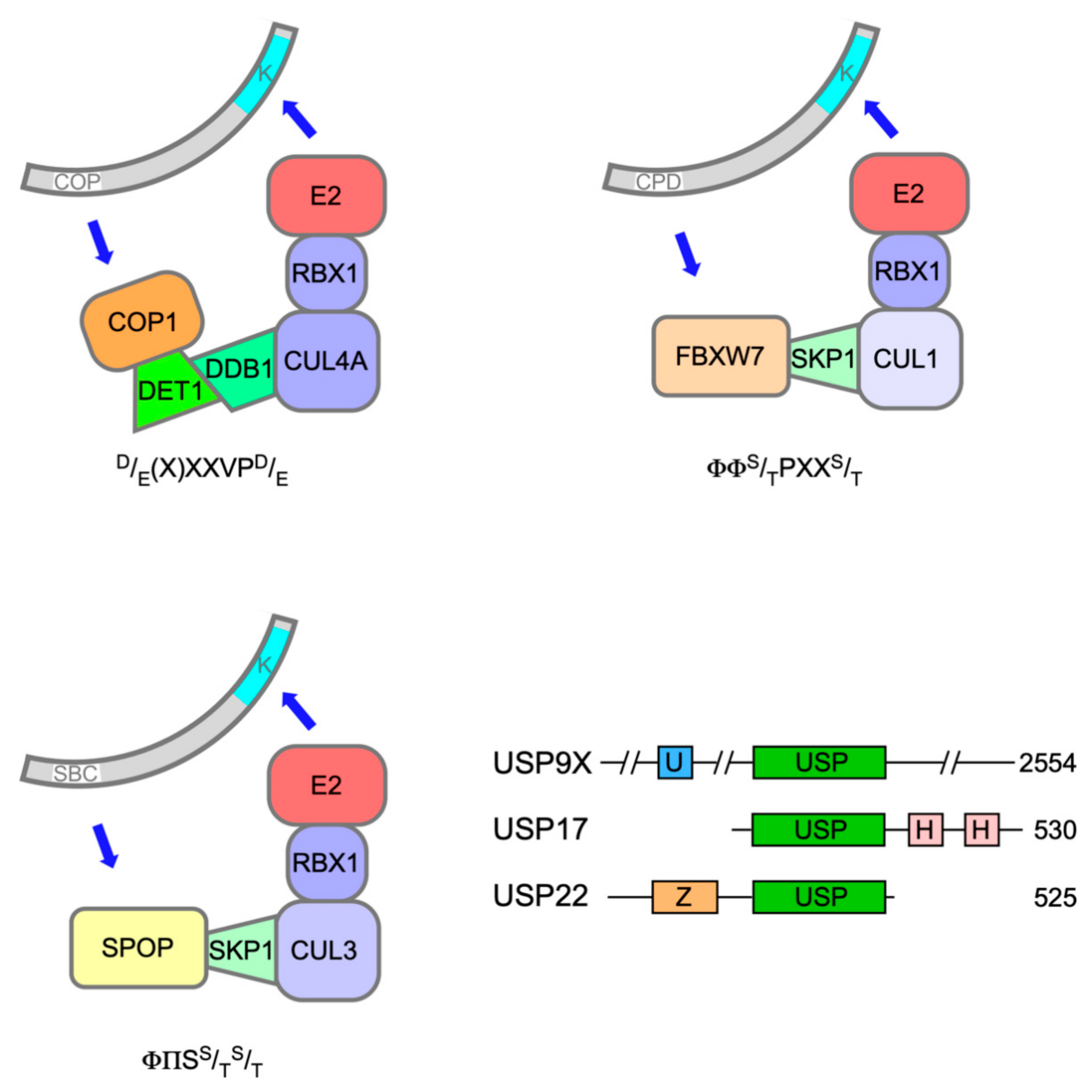
2. ETS-1, ETS-2 and the Constitutive Photomorphogenesis 1 (COP1) Complex
3. ERG Fusion Proteins and the Evasion of Ubiquitin-Mediated Proteolysis in Prostate Cancer
4. Interplay between PEA3 Relatives and COP1 during Insulin Secretion and in Cancer
5. TCFs Showcase Mono-Ubiquitination as a Non-Destructive Mode of Repression
6. TEL and the PNT Domain as Drivers of Turnover or Fusion Protein Activity
7. Discussion

{kind=link}
{kind=link}
| ETS Protein | E3 Ligase | DUB | References |
| ETS-1 | COP1 | USP9X | [30,35] |
| ETS-2 | COP1 | - | [35,40] |
| ERG | TRIM25, SPOP, FBXW7 | USP9X | [47,49,52,53,57] |
| ETV1/ER81 | COP1 | - | [72,73] |
| ETV4/PEA3/E1AF | COP1 | - | [72,73] |
| ETV5/ERM | COP1 | - | [72,73] |
| ETV6/TEL1 | FBXL6 | - | [98] |
| ETV7/TEL2 | FBXL6 | - | [98] |
| ELF3/ESE1 | FBXW1A | - | [105] |
| ELF4/MEF | SKP2 | - | [106] |
| PU.1/SPI-1 | FBXW7 | USP22 | [63,65] |
| ELK-1 | FBXO25 * | USP17 | [86,88,89] |
| ETS Fusion | E3 Ligase | DUB | References |
| TMPRSS2-ERG | TRIM25, FBXW7 | USP9X | [47,49,57] |
| EWS-FLI-1/ERGB | - | USP19 | [61] |
| ETV6/TEL1^-NTRK3 | RNF123 | - | [102] |
| ETV6/TEL1^-JAK2 | SOCS1 | - | [103] |
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemon, B.; Tjian, R. Orchestrated response: A symphony of transcription factors for gene control. Genes Dev. 2000, 14, 2551–2569. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.D.; Urness, L.D.; Thummel, C.S.; Klemsz, M.J.; McKercher, S.R. The ETS-domaln: A new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes Dev. 1990, 4, 1452–1453. [Google Scholar] [CrossRef] [PubMed]
- Leprince, D.; Gegonne, A.; Coll, J.; de Taisne, C.; Schneeberger, A.; Lagrou, C.; Stehelin, D. A putative second cell-derived oncogene of the avian leukaemia retrovirus E26. Nature 1983, 306, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R.; Nordheim, A. Gene regulation by Ets proteins. Biochim. Biophys. Acta BBA—Rev. Cancer 1993, 1155, 346–356. [Google Scholar] [CrossRef]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef]
- Mackereth, C.D.; Schärpf, M.; Gentile, L.N.; MacIntosh, S.E.; Slupsky, C.M.; McIntosh, L.P. Diversity in structure and function of the Ets family PNT domains. J. Mol. Biol. 2004, 342, 1249–1264. [Google Scholar] [CrossRef]
- Selvaraj, N.; Kedage, V.; Hollenhorst, P.C. Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells. Cell Commun. Signal. 2015, 13, 12. [Google Scholar] [CrossRef]
- Maroulakou, I.G.; Bowe, D.B. Expression and function of Ets transcription factors in mammalian development: A regulatory network. Oncogene 2000, 19, 6432–6442. [Google Scholar] [CrossRef]
- Hollenhorst, P.C. RAS/ERK pathway transcriptional regulation through ETS/AP-1 binding sites. Small GTPases 2012, 3, 154–158. [Google Scholar] [CrossRef][Green Version]
- Liu, M.; Gao, W.; Van Velkinburgh, J.C.; Wu, Y.; Ni, B.; Tian, Y. Role of Ets Proteins in Development, Differentiation, and Function of T-Cell Subsets. Med. Res. Rev. 2016, 36, 193–220. [Google Scholar] [CrossRef]
- Fry, E.A.; Mallakin, A.; Inoue, K. Translocations involving ETS family proteins in human cancer. Integr. Cancer Sci. Ther. 2018, 5, 1–12. [Google Scholar] [CrossRef]
- Hsing, M.; Wang, Y.; Rennie, P.S.; Cox, M.E.; Cherkasov, A. ETS transcription factors as emerging drug targets in cancer. Med. Res. Rev. 2020, 40, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-H.; Jaffray, E.; Hay, R.T.; Sharrocks, A.D. Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol. Cell 2003, 12, 63–74. [Google Scholar] [CrossRef]
- Guo, B.; Panagiotaki, N.; Warwood, S.; Sharrocks, A.D. Dynamic modification of the ETS transcription factor PEA3 by sumoylation and p300-mediated acetylation. Nucleic Acids Res. 2011, 39, 6303–6313. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell. Mol. Biol. Lett. 2021, 26, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Zagidullin, A.; Valiullina, A.; Sayarova, R.; Rizvanov, A. Small molecule modulators of RING-type E3 ligases: MDM and cullin families as targets. Front. Pharmacol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Garrett-Sinha, L.A. Review of Ets1 structure, function, and roles in immunity. Cell. Mol. Life Sci. 2013, 70, 3375–3390. [Google Scholar] [CrossRef]
- Lie-Venema, H.; Gittenberger-De Groot, A.C.; Van Empel, L.J.P.; Boot, M.J.; Kerkdijk, H.; De Kant, E.; DeRuiter, M.C. Ets-1 and Ets-2 transcription factors are essential for normal coronary and myocardial development in chicken embryos. Circ. Res. 2003, 92, 749–756. [Google Scholar] [CrossRef]
- Nie, S.; Bronner, M.E. Dual developmental role of transcriptional regulator Ets1 in Xenopus cardiac neural crest vs. heart mesoderm. Cardiovasc. Res. 2015, 106, 67–75. [Google Scholar] [CrossRef]
- Taveirne, S.; Wahlen, S.; van Loocke, W.; Kiekens, L.; Persyn, E.; van Ammel, E.; de Mulder, K.; Roels, J.; Tilleman, L.; Aumercier, M.; et al. The transcription factor ETS1 is an important regulator of human NK cell development and terminal differentiation. Blood 2020, 136, 288–298. [Google Scholar] [CrossRef]
- Ji, Z.; Degerny, C.; Vintonenko, N.; Deheuninck, J.; Foveau, B.; Leroy, C.; Coll, J.; Tulasne, D.; Baert, J.L.; Fafeur, V. Regulation of the Ets-1 transcription factor by sumoylation and ubiquitinylation. Oncogene 2007, 26, 395–406. [Google Scholar] [CrossRef]
- Potu, H.; Peterson, L.F.; Kandarpa, M.; Pal, A.; Sun, H.; Durham, A.; Harms, P.W.; Hollenhorst, P.C.; Eskiocak, U.; Talpaz, M.; et al. Usp9x regulates Ets-1 ubiquitination and stability to control NRAS expression and tumorigenicity in melanoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA 2018, 115, E1401–E1408. [Google Scholar] [CrossRef] [PubMed]
- Fulzele, A.; Bennett, E.J. Ubiquitin diGLY proteomics as an approach to identify and quantify the ubiquitin-modified proteome. Methods Mol. Biol. 2018, 1844, 363–384. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Vaessen, B.; Johnston, K. Structure of the Elk-1–DNA complex reveals how DNA- distal residues affect ETS domain recognition of DNA. Nat. Struct. Biol. 2000, 7, 3–8. [Google Scholar]
- Marine, J.C. Spotlight on the role of COP1 in tumorigenesis. Nat. Rev. Cancer 2012, 12, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zhang, Q.; Huang, Y.; Song, J.; Tomaino, R.; Ehrenberger, T.; Lim, E.; Liu, W.; Bronson, R.T.; Bowden, M.; et al. Phosphorylation of ETS1 by src family kinases prevents its recognition by the COP1 tumor suppressor. Cancer Cell 2014, 26, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Elia, A.E.H.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.C.; et al. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.C.; Chen, J.-R.; Lin, C.-H.; Zhang, G.; Lam, P.-S.; Wenger, K.H.; Mozaffari, F.B.; Huang, S.-T.; Borke, J.L. Tensile Strain-Induced Ets-2 Phosphorylation by CaMKII and the Homeostasis of Cranial Sutures. Plast. Reconstr. Surg. 2009, 123, 83S–93S. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Terashima, M.; Fukami, K.; Yamada, Y. PIASy controls ubiquitination-dependent proteasomal degradation of Ets-1. Biochem. J. 2007, 405, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Guen, V.J.; Gamble, C.; Flajolet, M.; Unger, S.; Thollet, A.; Ferandin, Y.; Superti-Furga, A.; Cohen, P.A.; Meijer, L.; Colas, P. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, 19525–19530. [Google Scholar] [CrossRef] [PubMed]
- Carrero, Z.I.; Kollareddy, M.; Chauhan, K.M.; Ramakrishnan, G.; Martinez, L.A. Mutant p53 protects ETS2 from non-canonical COP1/DET1 dependent degradation. Oncotarget 2016, 7, 12554–12567. [Google Scholar] [CrossRef]
- Bargonetti, J.; Prives, C. Gain-of-function mutant p53: History and speculation. J. Mol. Cell Biol. 2019, 11, 605–609. [Google Scholar] [CrossRef]
- Li, M.; Shin, Y.H.; Hou, L.; Huang, X.; Wei, Z.; Klann, E.; Zhang, P. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat. Cell Biol. 2008, 10, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Jolly, L.A.; Gecz, J.; Wood, S.A. La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 2015, 72, 2075–2089. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.J.; Rehn, M.; Hasemann, M.S.; Rapin, N.; Bagger, F.O.; Ohlsson, E.; Willer, A.; Frank, A.K.; Søndergaard, E.; Jendholm, J.; et al. ERG promotes the maintenance of hematopoietic stem cells by restricting their differentiation. Genes Dev. 2015, 29, 1915–1929. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, D.D.; Pharr, P.N.; Lavenburg, K.R.; Jackers, P.; Papas, T.S.; Ogawa, M.; Watson, D.K. Hemorrhage, Impaired Hematopoiesis, and Lethality in Mouse Embryos Carrying a Targeted Disruption of the Fli1Transcription Factor. Mol. Cell. Biol. 2000, 20, 5643–5652. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Srivastava, N.; Li, R.; Ravindranathan, P.; Hernandez, E.; Freeman, E.; Humphries, C.G.; Kapur, P.; Lotan, Y.; et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4251–4256. [Google Scholar] [CrossRef]
- Martín-Vicente, M.; Medrano, L.M.; Resino, S.; García-Sastre, A.; Martínez, I. TRIM25 in the regulation of the antiviral innate immunity. Front. Immunol. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef]
- Cheng, J.; Guo, J.; Wang, Z.; North, B.J.; Tao, K.; Dai, X.; Wei, W. Functional analysis of Cullin 3 E3 ligases in tumorigenesis. Biochim. Biophys. Acta BBA—Rev. Cancer 2018, 1869, 11–28. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Ren, S.; Murphy, S.J.; Dalangood, S.; Chang, C.; Pang, X.; Cui, Y.; Wang, L.; Pan, Y.; Zhang, X.; et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol. Cell 2015, 59, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Dai, X.; Lunardi, A.; Li, Z.; Inuzuka, H.; Liu, P.; Varmeh, S.; Zhang, J.; Cheng, L.; Sun, Y.; et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol. Cell 2015, 59, 917–930. [Google Scholar] [CrossRef]
- Shoag, J.; Liu, D.; Blattner, M.; Sboner, A.; Park, K.; Deonarine, L.; Robinson, B.D.; Mosquera, J.M.; Chen, Y.; Rubin, M.A.; et al. SPOP mutation drives prostate neoplasia without stabilizing oncogenic transcription factor ERG. J. Clin. Investig. 2018, 128, 381–386. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Zhang, W.; Ding, D.; Huang, Z.; Yan, Y.; Cao, W.; Pan, Y.; Hou, X.; Weroha, S.J.; Karnes, R.J.; et al. DNA Damage Promotes TMPRSS2-ERG Oncoprotein Destruction and Prostate Cancer Suppression via Signaling Converged by GSK3β and WEE1. Mol. Cell 2020, 79, 1008–1023.e4. [Google Scholar] [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; De Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Gierisch, M.E.; Pfistner, F.; Lopez-Garcia, L.A.; Harder, L.; Schäfer, B.W.; Niggli, F.K. Proteasomal degradation of the EWS-FLI1 fusion protein is regulated by a single lysine residue. J. Biol. Chem. 2016, 291, 26922–26933. [Google Scholar] [CrossRef]
- Gierisch, M.E.; Pedot, G.; Walser, F.; Lopez-Garcia, L.A.; Jaaks, P.; Niggli, F.K.; Schäfer, B.W. USP19 deubiquitinates EWS-FLI1 to regulate Ewing sarcoma growth. Sci. Rep. 2019, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.; Dakic, A.; Mifsud, S.; Di Rago, L.; Wu, L.; Nutt, S. Inactivation of PU.1 in adult mice leads to the development of myeloid leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Thacker, G.; Sharma, A.; Singh, A.K.; Upadhyay, V.; Sanyal, S.; Verma, S.P.; Tripathi, A.K.; Bhatt, M.L.B.; Trivedi, A.K. FBW7 Inhibits Myeloid Differentiation in Acute Myeloid Leukemia via GSK3-Dependent Ubiquitination of PU.1. Mol. Cancer Res. 2021, 19, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Suzuki, E.; Le Guezennec, X.; Stunnenberg, H.G.; Krasnov, A.; et al. A TFTC/STAGA Module Mediates Histone H2A and H2B Deubiquitination, Coactivates Nuclear Receptors, and Counteracts Heterochromatin Silencing. Mol. Cell 2008, 29, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Melo-Cardenas, J.; Xu, Y.; Wei, J.; Tan, C.; Kong, S.; Gao, B.; Montauti, E.; Kirsammer, G.; Licht, J.D.; Yu, J.; et al. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1-dependent mechanism. Blood 2018, 132, 423–434. [Google Scholar] [CrossRef]
- Oh, S.; Shin, S.; Janknecht, R. ETV1, 4 and 5: An oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta BBA—Rev. Cancer 2012, 1826, 1–12. [Google Scholar] [CrossRef]
- Herriges, J.C.; Verheyden, J.M.; Zhang, Z.; Sui, P.; Zhang, Y.; Anderson, M.J.; Swing, D.A.; Zhang, Y.; Lewandoski, M.; Sun, X. FGF-Regulated ETV Transcription Factors Control FGF-SHH Feedback Loop in Lung Branching. Dev. Cell 2015, 35, 322–332. [Google Scholar] [CrossRef]
- Takahashi, A.; Higashino, F.; Aoyagi, M.; Yoshida, K.; Itoh, M.; Kobayashi, M.; Totsuka, Y.; Kohgo, T.; Shindoh, M. E1AF degradation by a ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 2005, 327, 575–580. [Google Scholar] [CrossRef]
- Guo, B.; Sharrocks, A.D. Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase Signaling Initiates a Dynamic Interplay between Sumoylation and Ubiquitination To Regulate the Activity of the Transcriptional Activator PEA3. Mol. Cell. Biol. 2009, 29, 3204–3218. [Google Scholar] [CrossRef]
- Xiao, J.; Yang, S.; Shen, P.; Wang, Y.; Sun, H.; Ji, F.; Zhou, D. Phosphorylation of ETV4 at Ser73 by ERK kinase could block ETV4 ubiquitination degradation in colorectal cancer. Biochem. Biophys. Res. Commun. 2017, 486, 1062–1068. [Google Scholar] [CrossRef]
- Baert, J.L.; Beaudoin, C.; Monte, D.; Degerny, C.; Mauen, S.; De Launoit, Y. The 26S proteasome system degrades the ERM transcription factor and regulates its transcription-enhancing activity. Oncogene 2007, 26, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Baert, J.L.; Monte, D.; Verreman, K.; Degerny, C.; Coutte, L.; De Launoit, Y. The E3 ubiquitin ligase complex component COP1 regulates PEA3 group member stability and transcriptional activity. Oncogene 2010, 29, 1810–1820. [Google Scholar] [CrossRef]
- Vitari, A.C.; Leong, K.G.; Newton, K.; Yee, C.; O’Rourke, K.; Liu, J.; Phu, L.; Vij, R.; Ferrando, R.; Couto, S.S.; et al. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature 2011, 474, 403–406. [Google Scholar] [CrossRef]
- Ouyang, M.; Wang, H.; Ma, J.; Lü, W.; Li, J.; Yao, C.; Chang, G.; Bi, J.; Wang, S.; Wang, W. COP1, the negative regulator of ETV1, influences prognosis in triple-negative breast cancer. BMC Cancer 2015, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Cao, Z.; Wong, E.W.P.; Guan, Y.; Ma, W.; Zhang, J.Q.; Walczak, E.G.; Murphy, D.; Ran, L.; Sirota, I.; et al. COP1/DET1/ETS axis regulates ERK transcriptome and sensitivity to MAPK inhibitors. J. Clin. Investig. 2018, 128, 1442–1457. [Google Scholar] [CrossRef]
- Suriben, R.; Kaihara, K.A.; Paolino, M.; Reichelt, M.; Kummerfeld, S.K.; Modrusan, Z.; Dugger, D.L.; Newton, K.; Sagolla, M.; Webster, J.D.; et al. β-Cell Insulin Secretion Requires the Ubiquitin Ligase COP1. Cell 2015, 163, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dugger, D.L.; Sengupta-Ghosh, A.; Ferrando, R.E.; Chu, F.; Tao, J.; Lam, W.; Haller, S.; Chan, S.; Sa, S.; et al. Ubiquitin ligase COP1 coordinates transcriptional programs that control cell type specification in the developing mouse brain. Proc. Natl. Acad. Sci. USA 2018, 115, 11244–11249. [Google Scholar] [CrossRef] [PubMed]
- Zhanga, Y.; Yokoyamaa, S.; Herrigesa, J.C.; Zhanga, Z.; Younga, R.E.; Verheydena, J.M.; Suna, X. E3 ubiquitin ligase RFWD2 controls lung branching through protein-level regulation of ETV transcription factors. Proc. Natl. Acad. Sci. USA 2016, 113, 7557–7562. [Google Scholar] [CrossRef] [PubMed]
- Marais, R.; Wynne, J.; Treisman, R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393. [Google Scholar] [CrossRef]
- Janknecht, R.; Ernst, W.H.; Pingoud, V.; Nordheim, A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 1993, 12, 5097–5104. [Google Scholar] [CrossRef]
- Gille, H.; Kortenjann, M.; Thomae, O.; Moomaw, C.; Slaughter, C.; Cobb, M.H.; Shaw, P.E. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995, 14, 951–962. [Google Scholar] [CrossRef]
- Nentwich, O.; Dingwell, K.S.; Nordheim, A.; Smith, J.C. Downstream of FGF during mesoderm formation in Xenopus: The roles of Elk-1 and Egr-1. Dev. Biol. 2009, 336, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Saxton, J.; Ferjentsik, Z.; Ducker, C.; Johnson, A.D.; Shaw, P.E. Stepwise evolution of Elk-1 in early deuterostomes. FEBS J. 2016, 283, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Xie, B.; Adler, V.; Fried, V.A.; Davis, R.J.; Ronai, Z. C-jun NH2-terminal kinases target the ubiquitination of their associated transcription factors. J. Biol. Chem. 1997, 272, 32163–32168. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.L.; Saxton, J.; Shelton, S.J.; Begitt, A.; Holliday, N.D.; Hipskind, R.A.; Shaw, P.E. Dimer formation and conformational flexibility ensure cytoplasmic stability and nuclear accumulation of Elk-1. Nucleic Acids Res. 2011, 39, 6390–6402. [Google Scholar] [CrossRef]
- Ducker, C.; Chow, L.K.Y.; Saxton, J.; Handwerger, J.; McGregor, A.; Strahl, T.; Layfield, R.; Shaw, P.E. De-ubiquitination of ELK-1 by USP17 potentiates mitogenic gene expression and cell proliferation. Nucleic Acids Res. 2019, 47, 4495–4508. [Google Scholar] [CrossRef] [PubMed]
- Hagens, O.; Minina, E.; Schweiger, S.; Ropers, H.H.; Kalscheuer, V. Characterization of FBX25, encoding a novel brain-expressed F-box protein. Biochim. Biophys. Acta BBA—Gen. Subj. 2006, 1760, 110–118. [Google Scholar] [CrossRef]
- Teixeira, F.R.; Manfiolli, A.O.; Soares, C.S.; Baqui, M.M.A.; Koide, T.; Gomes, M.D. The F-box protein FBXO25 promotes the proteasome-dependent degradation of ELK-1 protein. J. Biol. Chem. 2013, 288, 28152–28162. [Google Scholar] [CrossRef]
- Quintero-Barceinas, R.S.; Gehringer, F.; Ducker, C.; Saxton, J.; Shaw, P.E. ELK-1 ubiquitination status and transcriptional activity are modulated independently of F-Box protein FBXO25. J. Biol. Chem. 2021, 296, 100214. [Google Scholar] [CrossRef] [PubMed]
- Ducker, C.; Shaw, P.E. USP17-mediated de-ubiquitination and cancer: Clients cluster around the cell cycle. Int. J. Biochem. Cell Biol. 2021, 130, 105886. [Google Scholar] [CrossRef]
- Rosati, R.; Patki, M.; Chari, V.; Dakshnamurthy, S.; McFall, T.; Saxton, J.; Kidder, B.L.; Shaw, P.E.; Ratnam, M. The amino-terminal domain of the androgen receptor co-opts extracellular signal-regulated kinase (ERK) docking sites in ELK1 protein to induce sustained gene activation that supports prostate cancer cell growth. J. Biol. Chem. 2016, 291, 25983–25998. [Google Scholar] [CrossRef]
- Rosati, R.; Polin, L.; Ducker, C.; Li, J.; Bao, X.; Selvakumar, D.; Kim, S.; Xhabija, B.; Larsen, M.; McFall, T.; et al. Strategy for tumor-selective disruption of androgen receptor function in the spectrum of prostate cancer. Clin. Cancer Res. 2018, 24, 6509–6522. [Google Scholar] [CrossRef] [PubMed]
- Pardy, L.; Rosati, R.; Soave, C.; Huang, Y.; Kim, S.; Ratnam, M. The ternary complex factor protein ELK1 is an independent prognosticator of disease recurrence in prostate cancer. Prostate 2020, 80, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Buchwalter, G.; Dubois-Pot, H.; Cler, E.; Zheng, H.; Wasylyk, B. The Ternary Complex Factor Net Is Downregulated by Hypoxia and Regulates Hypoxia-Responsive Genes. Mol. Cell. Biol. 2007, 27, 4133–4141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hooker, E.; Baldwin, C.; Roodman, V.; Batra, A.; Isa, N.N.; Takano, T.; Lemay, S. Binding and inhibition of the ternary complex factor Elk-4/Sap1 by the adapter protein Dok-4. Biochem. J. 2017, 474, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Rasighaemi, P.; Ward, A.C. ETV6 and ETV7: Siblings in hematopoiesis and its disruption in disease. Crit. Rev. Oncol. Hematol. 2017, 116, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.A.; Phillips, M.L.; Kim, W.; Gingery, M.; Tran, H.H.; Robinson, M.A.; Faham, S.; Bowie, J.U. Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 2001, 20, 4173–4182. [Google Scholar] [CrossRef] [PubMed]
- Roukens, M.G.; Alloul-Ramdhani, M.; Moghadasi, S.; Op den Brouw, M.; Baker, D.A. Downregulation of Vertebrate Tel (ETV6) and Drosophila Yan Is Facilitated by an Evolutionarily Conserved Mechanism of F-Box-Mediated Ubiquitination. Mol. Cell. Biol. 2008, 28, 4394–4406. [Google Scholar] [CrossRef] [PubMed]
- Roukens, M.G.; Alloul-Ramdhani, M.; Vertegaal, A.C.O.; Anvarian, Z.; Balog, C.I.A.; Deelder, A.M.; Hensbergen, P.J.; Baker, D.A. Identification of a New Site of Sumoylation on Tel (ETV6) Uncovers a PIAS-Dependent Mode of Regulating Tel Function. Mol. Cell. Biol. 2008, 28, 2342–2357. [Google Scholar] [CrossRef]
- Molina, M.D.; Quirin, M.; Haillot, E.; De Crozé, N.; Range, R.; Rouel, M.; Jimenez, F.; Amrouche, R.; Chessel, A.; Lepage, T. MAPK and GSK3/ß-TRCP-mediated degradation of the maternal Ets domain transcriptional repressor Yan/Tel controls the spatial expression of nodal in the sea urchin embryo. PLoS Genet. 2018, 14, e1007621. [Google Scholar] [CrossRef]
- Lannon, C.L.; Sorensen, P.H.B. ETV6-NTRK3: A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 2005, 15, 215–223. [Google Scholar] [CrossRef]
- Tognon, C.E.; Rafn, B.; Cetinbas, N.M.; Kamura, T.; Trigo, G.; Rotblat, B.; Okumura, F.; Matsumoto, M.; Chow, C.; Davare, M.; et al. Insulin-like growth factor 1 receptor stabilizes the ETV6-NTRK3 chimeric oncoprotein by blocking its KPC1/Rnf123-mediated proteasomal degradation. J. Biol. Chem. 2018, 293, 12502–12515. [Google Scholar] [CrossRef] [PubMed]
- Kamizono, S.; Hanada, T.; Yasukawa, H.; Minoguchi, S.; Kato, R.; Minoguchi, M.; Hattori, K.; Hatakeyama, S.; Yada, M.; Morita, S.; et al. The SOCS Box of SOCS-1 Accelerates Ubiquitin-dependent Proteolysis of TEL-JAK2. J. Biol. Chem. 2001, 276, 12530–12538. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010, 29, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Rayala, S.K.; Kumar, R. Phosphorylation-dependent regulation of stability and transforming potential of ETS transcriptional factor ESE-1 by p21-activated kinase. J. Biol. Chem. 2007, 282, 19820–19830. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hedvat, C.V.; Mao, S.; Zhu, X.-H.; Yao, J.; Nguyen, H.; Koff, A.; Nimer, S.D. The ETS Protein MEF Is Regulated by Phosphorylation-Dependent Proteolysis via the Protein-Ubiquitin Ligase SCFSkp2. Mol. Cell. Biol. 2006, 26, 3114–3123. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.E.; Paccez, J.D.; Duncan, K.C.; Morale, M.G.; Simabuco, F.M.; Dillon, S.; Correa, R.G.; Gu, X.; Libermann, T.A.; Zerbini, L.F. GADD45α and γ interaction with CDK11p58 regulates SPDEF protein stability and SPDEF-mediated effects on cancer cell migration. Oncotarget 2016, 7, 13865–13879. [Google Scholar] [CrossRef][Green Version]
- Guo, J.C.; Yang, Y.J.; Guo, M.; Zhang, J.Q.; Zheng, J.F.; Liu, Z. Involvement of CDK11B-mediated SPDEF ubiquitination and SPDEF-mediated microRNA-448 activation in the oncogenicity and self-renewal of hepatocellular carcinoma stem cells. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef]
- Lumpkin, R.J.; Gu, H.; Zhu, Y.; Leonard, M.; Ahmad, A.S.; Clauser, K.R.; Meyer, J.G.; Bennett, E.J.; Komives, E.A. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. Ubisite approach for comprehensive mapping of lysine and n-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.P.; et al. Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Jones, D.A.; Graves, B.J. Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 2004, 32, 5693–5702. [Google Scholar] [CrossRef] [PubMed]


| ETS Protein | ETS Domain Ubiquitination Site | References |
|---|---|---|
| ETS-1 | K377, K388, K399, K404 | [30,109,110,111,112] |
| ETS-2 | K416, K427, K432 | [109,110,111,112] |
| FLI-1/ERGB | K334 (K380 EWS-FLI) | [60,112] |
| ETV2/ER71 | K294 | [109,110,112] |
| ETV6/TEL1 | K393, K403 | [112,113] |
| ETV7/TEL2 | K293 | [114] |
| GABPα | K359, K366, K373 | [109,110,112,113,114,115,116] |
| ELF1 | K226, K244 | [109,110,112] |
| ELF2/NERF | K290 | [110] |
| ELF3/ESE1 | K294, K328 | [115] |
| ELK-1 | K35, K52, K59 | [86] |
| ELK-3/NET | K83 | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ducker, C.; Shaw, P.E. Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development. Int. J. Mol. Sci. 2021, 22, 5119. https://doi.org/10.3390/ijms22105119
Ducker C, Shaw PE. Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development. International Journal of Molecular Sciences. 2021; 22(10):5119. https://doi.org/10.3390/ijms22105119
Chicago/Turabian StyleDucker, Charles, and Peter E. Shaw. 2021. "Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development" International Journal of Molecular Sciences 22, no. 10: 5119. https://doi.org/10.3390/ijms22105119
APA StyleDucker, C., & Shaw, P. E. (2021). Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development. International Journal of Molecular Sciences, 22(10), 5119. https://doi.org/10.3390/ijms22105119