Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples

 , ,
, ,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
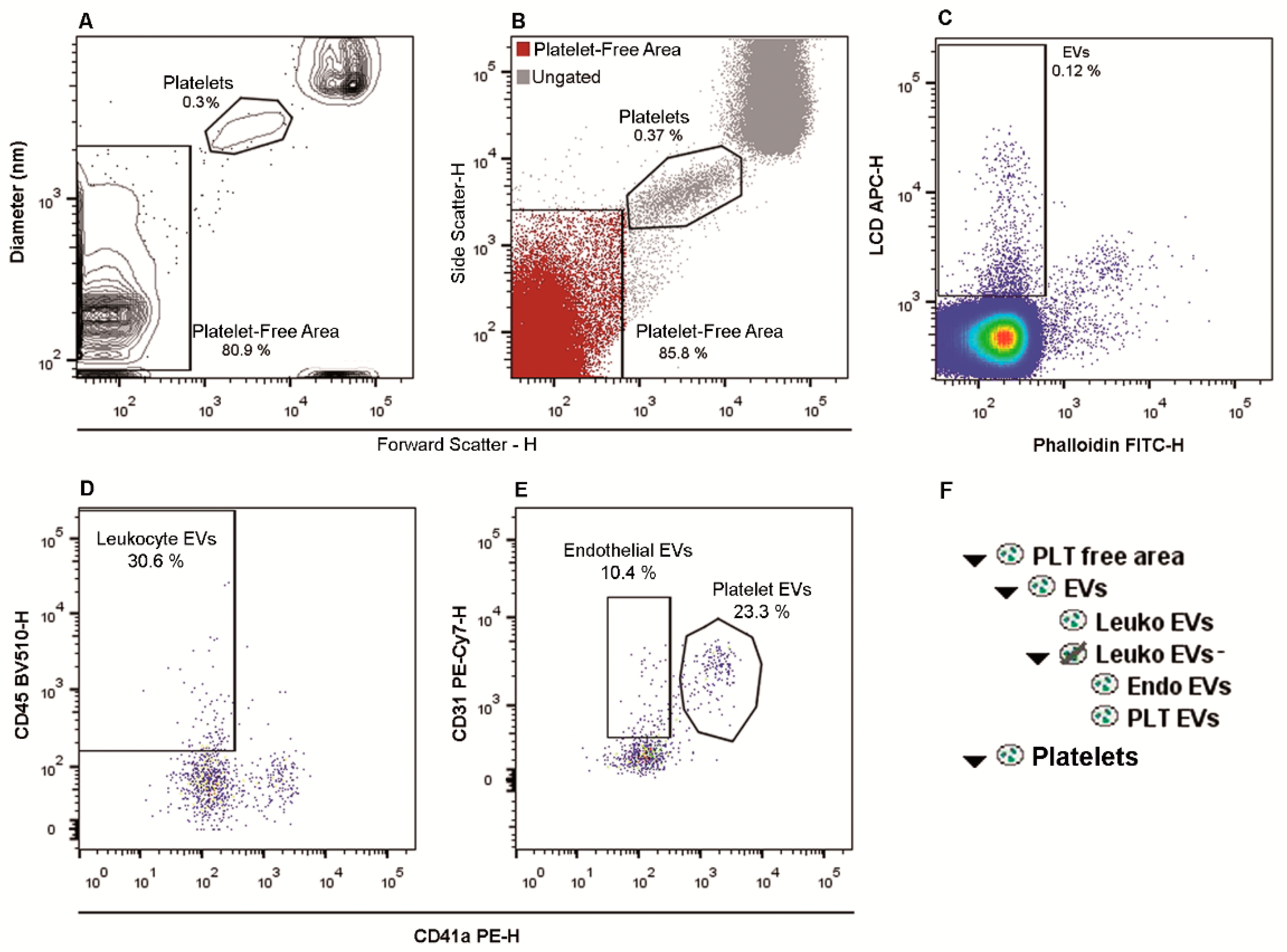
2.1. Flow Cytometry Identification and Subtyping of EVs from Human Whole Peripheral Blood Samples
2.2. Sensitivity of the Flow Cytometry Fluorescence Triggering
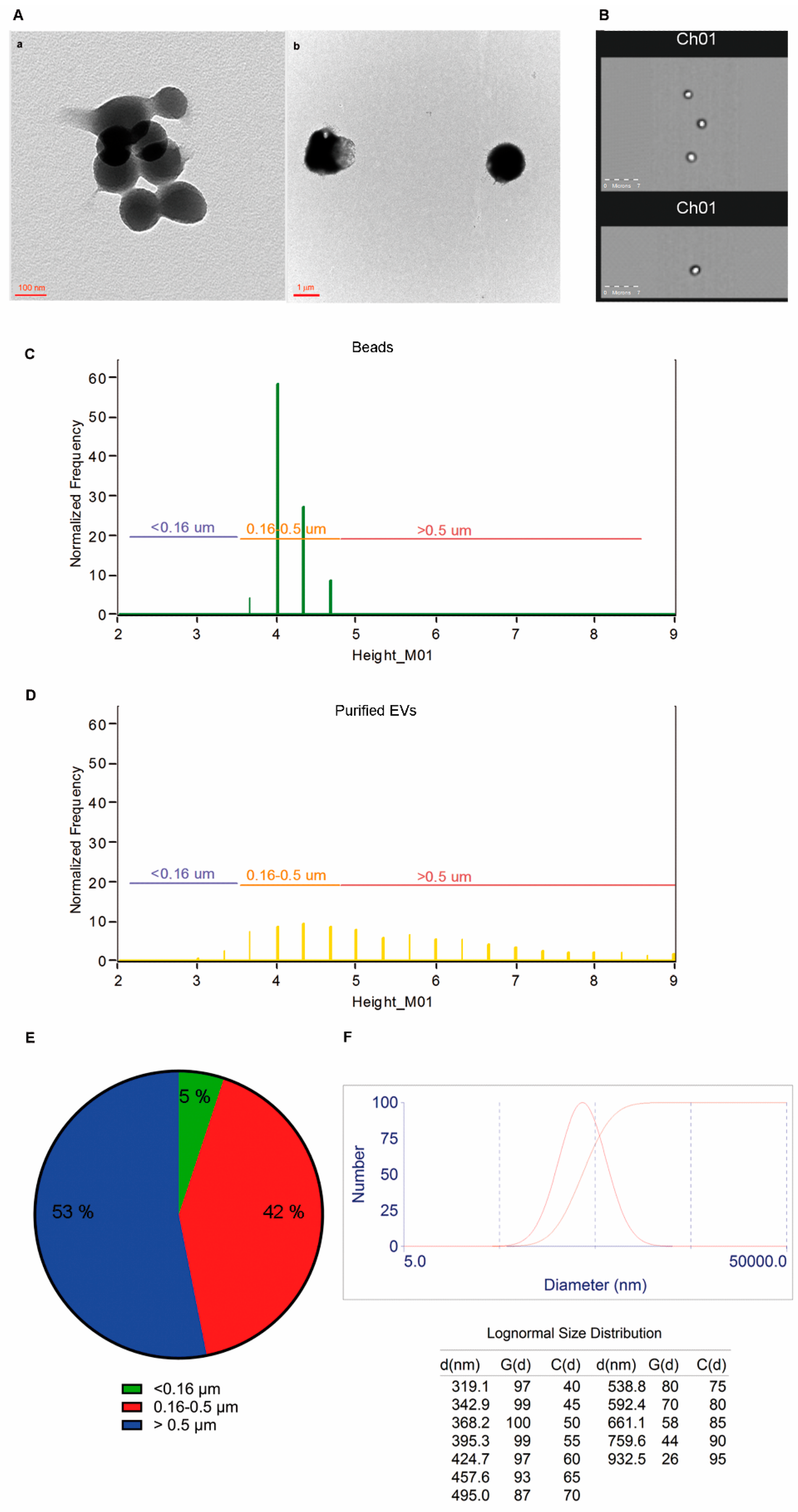
2.3. Size and Morphology of LCD Positive Events
2.4. LCD Stains EVs through A Dual Action Mechanism
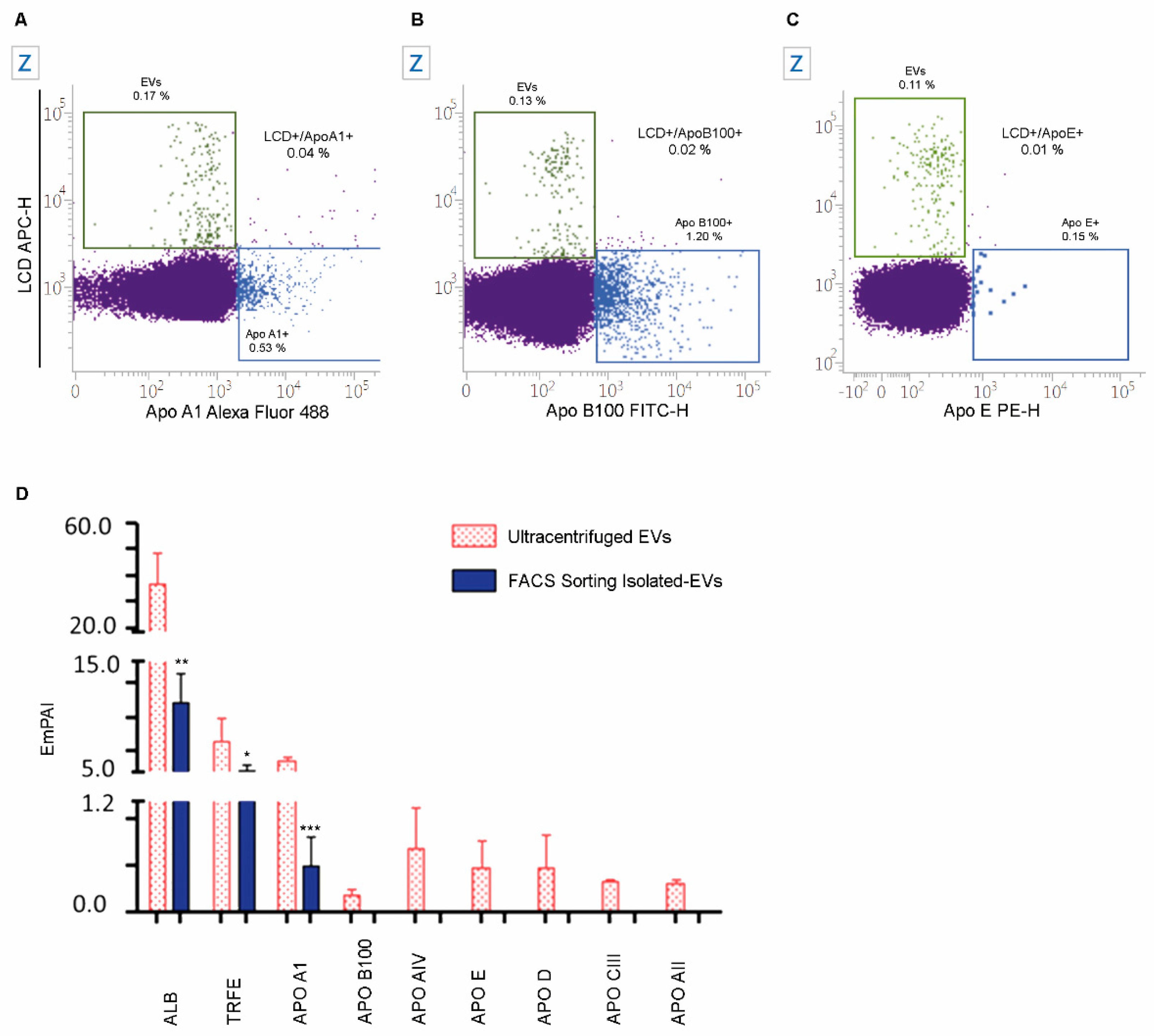
2.5. The Specificity of LCD Staining
2.6. EVs Numbers Obtained by Applying the LCD-Based or the Platelet-Free-Plasma (PFP) Protocol
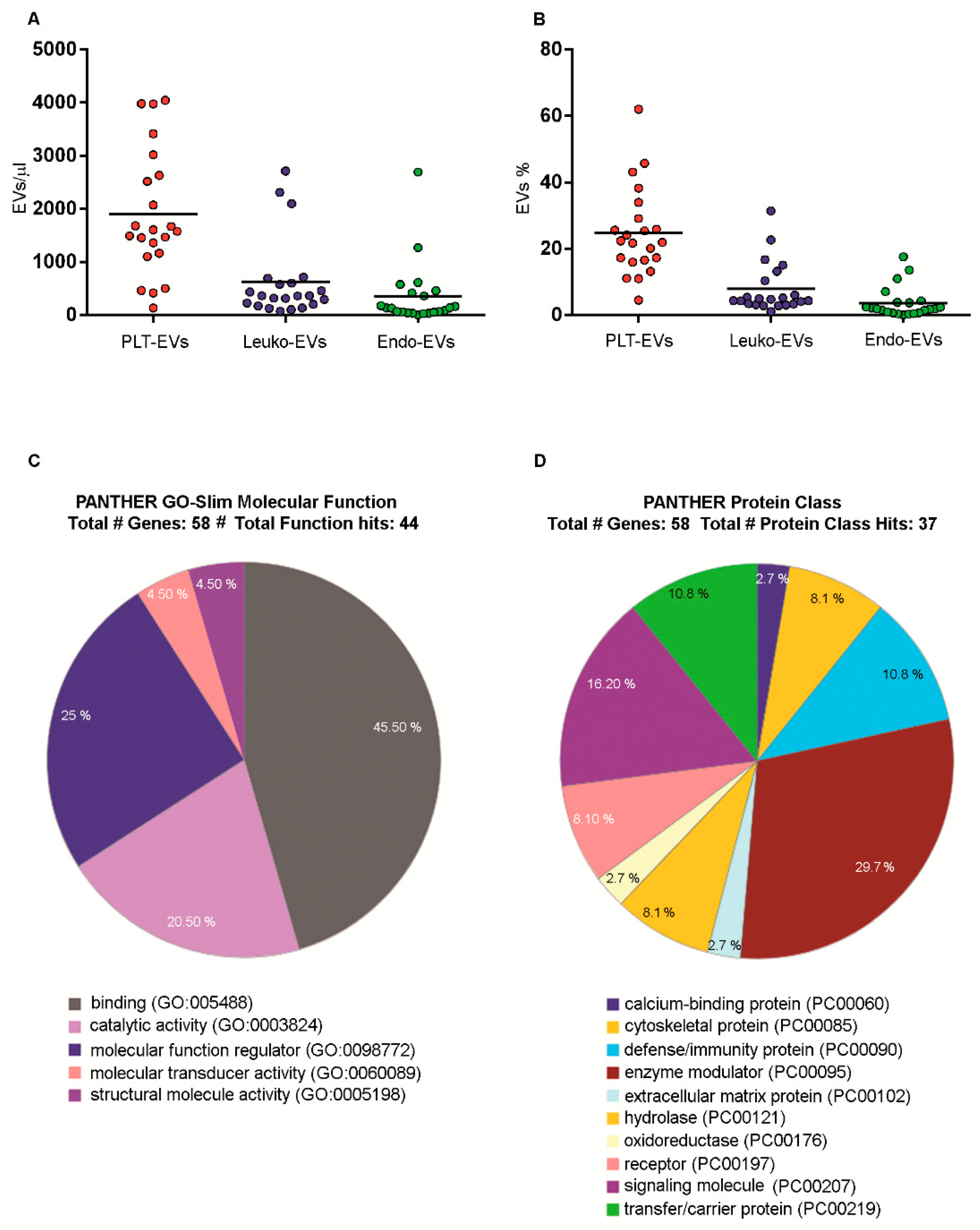
2.7. Numbers and Cargo of EVs Isolated from PB Samples of Healthy Volunteers
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Flow Cytometry EVs Staining
4.2.1. Analysis of Platelet Activation
4.2.2. Staining of Apolipoproteins
4.3. Flow Cytometry Extracellular Vesicle Acquisition and Analysis
4.4. Synthesis and Staining of Rhodamine-Liposomes
4.5. Carbonyl Cyanide 3-Chlorophenylhydrazone Impact on LCD Staining of Extracellular Vesicles
4.6. EVs Separation by Fluorescence-Activated Cell Sorting
4.7. Transmission Electron Microscopy Analysis
4.8. Dynamic Laser Light Scattering (DLS) Analysis of Purified EVs
4.9. ImageStream Analysis of EVs
4.10. EVs Label-Free Proteomics
4.11. Statistical Analysis.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Allophycocyanin |
| BV | Brilliant Violet |
| CCCP | Carbonyl cyanide 3-chlorophenylhydrazone |
| DMSO | Dimethyl Sulfoxide |
| DLS | Dynamic Light Scattering |
| emPAI | Exponentially Modified Protein Abundance Index |
| ERF | Equivalent reference fluorophores |
| EVs | Extracellular vesicles |
| FC | Flow cytometry |
| FSC | Forward scatter |
| FITC | Fluorescein isothiocyanate |
| FMO | Fluorescence Minus One |
| LCD | Lipophilic Cationic Dye |
| PB | Peripheral Blood |
| PBS | Phosphate-buffered saline |
| PE | Phycoerythrin |
| PE-Cy7 | Phycoerythrin-Cyanine 7 |
| PerCP-Cy5.5 | Peridinin Chlorophyll Protein- Cyanine 5.5 |
| PFC | Polychromatic flow cytometry |
| PFP | Platelet-Free Plasma |
| SSC | Side scatter |
| TEM | Transmission Electron Microscopy |
| UC | Ultracentrifugation |
References
- Shantsila, E.; Montoro-García, S.; Gallego, P.; Lip, G.Y.H. Circulating microparticles: Challenges and perspectives of flow cytometric assessment. Thromb. Haemost. 2014, 111, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Buca, D.; Bologna, G.; D’Amico, A.; Cugini, S.; Musca, F.; Febbo, M.; D’Arcangelo, D.; Buca, D.; Simeone, P.; Liberati, M.; et al. Extracellular Vesicles in Feto-Maternal Crosstalk and Pregnancy Disorders. Int. J. Mol. Sci. 2020, 21, 2120. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Bologna, G.; Lanuti, P.; Pierdomenico, L.; Guagnano, M.T.; Pieragostino, D.; Del Boccio, P.; Vergara, D.; Marchisio, M.; Miscia, S.; et al. Extracellular Vesicles as Signaling Mediators and Disease Biomarkers across Biological Barriers. Int. J. Mol. Sci. 2020, 21, 2514. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.R.; Bebawy, M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef] [PubMed]
- Karasu, E.; Eisenhardt, S.U.; Harant, J.; Huber-Lang, M. Extracellular Vesicles: Packages Sent With Complement. Front. Immunol. 2018, 9, 721. [Google Scholar] [CrossRef]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef]
- Tramontano, A.F.; Lyubarova, R.; Tsiakos, J.; Palaia, T.; Deleon, J.R.; Ragolia, L. Circulating endothelial microparticles in diabetes mellitus. Mediat. Inflamm. 2010, 2010, 250476. [Google Scholar] [CrossRef]
- Lanuti, P.; Santilli, F.; Marchisio, M.; Pierdomenico, L.; Vitacolonna, E.; Santavenere, E.; Iacone, A.; Davì, G.; Romano, M.; Miscia, S. A novel flow cytometric approach to distinguish circulating endothelial cells from endothelial microparticles: Relevance for the evaluation of endothelial dysfunction. J. Immunol. Methods 2012, 380, 16–22. [Google Scholar] [CrossRef]
- Pieragostino, D.; Cicalini, I.; Lanuti, P.; Ercolino, E.; di Ioia, M.; Zucchelli, M.; Zappacosta, R.; Miscia, S.; Marchisio, M.; Sacchetta, P.; et al. Enhanced release of acid sphingomyelinase-enriched exosomes generates a lipidomics signature in CSF of Multiple Sclerosis patients. Sci. Rep. 2018, 8, 3071. [Google Scholar] [CrossRef]
- Ciardiello, C.; Leone, A.; Lanuti, P.; Roca, M.S.; Moccia, T.; Minciacchi, V.R.; Minopoli, M.; Gigantino, V.; De Cecio, R.; Rippa, M.; et al. Large oncosomes overexpressing integrin alpha-V promote prostate cancer adhesion and invasion via AKT activation. J. Exp. Clin. Cancer Res. 2019, 38, 317. [Google Scholar] [CrossRef]
- Ciccocioppo, F.; Lanuti, P.; Centonze, D.; Miscia, S.; Marchisio, M. The Link Among Neurological Diseases: Extracellular Vesicles as a Possible Brain Injury Footprint. Neurosignals 2019, 27, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; Cassidy, H.; Ní Áinle, F.; Caprodossi, A.; Lanuti, P.; et al. Platelet-Derived Microparticles From Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk With Cancer and Endothelial Cells. Front. Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Marchisio, M.; Lanuti, P.; Boccatonda, A.; Miscia, S.; Davì, G. Microparticles as new markers of cardiovascular risk in diabetes and beyond. Thromb. Haemost. 2016, 116, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Brocco, D.; Lanuti, P.; Simeone, P.; Bologna, G.; Pieragostino, D.; Cufaro, M.C.; Graziano, V.; Peri, M.; Di Marino, P.; De Tursi, M.; et al. Circulating Cancer Stem Cell-Derived Extracellular Vesicles as a Novel Biomarker for Clinical Outcome Evaluation. J. Oncol. 2019, 2019, 5879616. [Google Scholar] [CrossRef] [PubMed]
- Cufaro, M.C.; Pieragostino, D.; Lanuti, P.; Rossi, C.; Cicalini, I.; Federici, L.; De Laurenzi, V.; Del Boccio, P. Extracellular Vesicles and Their Potential Use in Monitoring Cancer Progression and Therapy: The Contribution of Proteomics. J. Oncol. 2019, 2019, 1639854. [Google Scholar] [CrossRef] [PubMed]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- van der Pol, E.; Coumans, F.; Varga, Z.; Krumrey, M.; Nieuwland, R. Innovation in detection of microparticles and exosomes. J. Thromb. Haemost. 2013, 11, 36–45. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Mullier, F.; Bailly, N.; Chatelain, C.; Chatelain, B.; Dogné, J.-M. Pre-analytical issues in the measurement of circulating microparticles: Current recommendations and pending questions. J. Thromb. Haemost. 2013, 11, 693–696. [Google Scholar] [CrossRef]
- Yuana, Y.; Bertina, R.M.; Osanto, S. Pre-analytical and analytical issues in the analysis of blood microparticles. Thromb. Haemost. 2011, 105, 396–408. [Google Scholar] [CrossRef]
- Lee, R.D.; Barcel, D.A.; Williams, J.C.; Wang, J.G.; Boles, J.C.; Manly, D.A.; Key, N.S.; Mackman, N. Pre-analytical and analytical variables affecting the measurement of plasma-derived microparticle tissue factor activity. Thromb. Res. 2012, 129, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Chandler, W.L.; Yeung, W.; Tait, J.F. A new microparticle size calibration standard for use in measuring smaller microparticles using a new flow cytometer. J. Thromb. Haemost. 2011, 9, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Celia, C.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Cilurzo, F.; Grande, R.; Diomede, F.; Vespa, S.; Canonico, B.; et al. Diameters and Fluorescence Calibration for Extracellular Vesicle Analyses by Flow Cytometry. Int. J. Mol. Sci. 2020, 21, 7885. [Google Scholar] [CrossRef] [PubMed]
- Stoner, S.A.; Duggan, E.; Condello, D.; Guerrero, A.; Turk, J.R.; Narayanan, P.K.; Nolan, J.P. High sensitivity flow cytometry of membrane vesicles. Cytometry. A 2016, 89, 196–206. [Google Scholar] [CrossRef]
- He, Z.; Tang, Y.; Qin, C. Increased circulating leukocyte-derived microparticles in ischemic cerebrovascular disease. Thromb. Res. 2017, 154, 19–25. [Google Scholar] [CrossRef]
- Zarà, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. [Google Scholar] [CrossRef]
- Sódar, B.W.; Kittel, Á.; Pálóczi, K.; Vukman, K.V.; Osteikoetxea, X.; Szabó-Taylor, K.; Németh, A.; Sperlágh, B.; Baranyai, T.; Giricz, Z.; et al. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Sci. Rep. 2016, 6, 24316. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 1–29. [Google Scholar] [CrossRef]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef]
- Poncelet, P.; Robert, S.; Bouriche, T.; Bez, J.; Lacroix, R.; Dignat-George, F. Standardized counting of circulating platelet microparticles using currently available flow cytometers and scatter-based triggering: Forward or side scatter? Cytometry. A 2016, 89, 148–158. [Google Scholar] [CrossRef]
- Lacroix, R.; Judicone, C.; Mooberry, M.; Boucekine, M.; Key, N.S.; Dignat-George, F.; The ISTH SSC Workshop. Standardization of pre-analytical variables in plasma microparticle determination: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J. Thromb. Haemost. 2013, 11, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Judicone, C.; Poncelet, P.; Robert, S.; Arnaud, L.; Sampol, J.; Dignat-George, F. Impact of pre-analytical parameters on the measurement of circulating microparticles: Towards standardization of protocol. J. Thromb. Haemost. 2012, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Cointe, S.; Judicone, C.; Robert, S.; Mooberry, M.J.; Poncelet, P.; Wauben, M.; Nieuwland, R.; Key, N.S.; Dignat-George, F.; Lacroix, R. Standardization of microparticle enumeration across different flow cytometry platforms: Results of a multicenter collaborative workshop. J. Thromb. Haemost. 2017, 15, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.C.; Tual-Chalot, S.; Leonetti, D.; Andriantsitohaina, R. Microparticles: Targets and tools in cardiovascular disease. Trends Pharmacol. Sci. 2011, 32, 659–665. [Google Scholar] [CrossRef]
- Buzas, E.I.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- van der Pol, E.; Sturk, A.; van Leeuwen, T.; Nieuwland, R.; Coumans, F.; ISTH-SSC-VB Working group. Standardization of extracellular vesicle measurements by flow cytometry through vesicle diameter approximation. J. Thromb. Haemost. 2018, 16, 1236–1245. [Google Scholar] [CrossRef]
- Dey-Hazra, E.; Hertel, B.; Kirsch, T.; Woywodt, A.; Lovric, S.; Haller, H.; Haubitz, M.; Erdbruegger, U. Detection of circulating microparticles by flow cytometry: Influence of centrifugation, filtration of buffer, and freezing. Vasc. Health Risk Manag. 2010, 6, 1125–1133. [Google Scholar] [CrossRef]
- Chandler, W.L. Microparticle counts in platelet-rich and platelet-free plasma, effect of centrifugation and sample-processing protocols. Blood Coagul. Fibrinolysis 2013, 24, 125–132. [Google Scholar] [CrossRef]
- Chattopadhyay, P.K.; Hogerkorp, C.-M.; Roederer, M. A chromatic explosion: The development and future of multiparameter flow cytometry. Immunology 2008, 125, 441–449. [Google Scholar] [CrossRef]
- Lee, J.A.; Spidlen, J.; Boyce, K.; Cai, J.; Crosbie, N.; Dalphin, M.; Furlong, J.; Gasparetto, M.; Goldberg, M.; Goralczyk, E.M.; et al. MIFlowCyt: The minimum information about a Flow Cytometry Experiment. Cytometry. A 2008, 73, 926–930. [Google Scholar] [CrossRef]
- Welsh, J.A.; Van Der Pol, E.; Arkesteijn, G.J.A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles 2020, 9, 1713526. [Google Scholar] [CrossRef] [PubMed]
- Stagnara, J.; Garnache Ottou, F.; Angelot, F.; Mourey, G.; Seilles, E.; Biichlé, S.; Saas, P.; Racadot, E. Correlation between platelet-derived microparticle enumeration by flow cytometry and phospholipid-dependent procoagulant activity in microparticles: The centrifugation step matters! Thromb. Haemost. 2012, 107, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Dignat-George, F.; Freyssinet, J.-M.; Key, N.S. Centrifugation is a crucial step impacting microparticle measurement. Platelets 2009, 20, 225–226. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shah, M.D.; Bergeron, A.L.; Dong, J.-F.; López, J.A. Flow cytometric measurement of microparticles: Pitfalls and protocol modifications. Platelets 2008, 19, 365–372. [Google Scholar] [CrossRef]
- Christersson, C.; Lindahl, B.; Siegbahn, A. The composition and daily variation of microparticles in whole blood in stable coronary artery disease. Scand. J. Clin. Lab. Investig. 2016, 76, 25–32. [Google Scholar] [CrossRef]
- Christersson, C.; Johnell, M.; Siegbahn, A. Evaluation of microparticles in whole blood by multicolour flow cytometry assay. Scand. J. Clin. Lab. Investig. 2013, 73, 229–239. [Google Scholar] [CrossRef]
- Mobarrez, F.; Antovic, J.; Egberg, N.; Hansson, M.; Jörneskog, G.; Hultenby, K.; Wallén, H. A multicolor flow cytometric assay for measurement of platelet-derived microparticles. Thromb. Res. 2010, 125, e110–e116. [Google Scholar] [CrossRef]
- Cossarizza, A.; Chang, H.-D.; Radbruch, A.; Acs, A.; Adam, D.; Adam-Klages, S.; Agace, W.W.; Aghaeepour, N.; Akdis, M.; Allez, M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019, 49, 1457–1973. [Google Scholar] [CrossRef]
- Lanuti, P.; Simeone, P.; Rotta, G.; Almici, C.; Avvisati, G.; Azzaro, R.; Bologna, G.; Budillon, A.; Di Cerbo, M.; Di Gennaro, E.; et al. A standardized flow cytometry network study for the assessment of circulating endothelial cell physiological ranges. Sci. Rep. 2018, 8, 5823. [Google Scholar] [CrossRef]
- Streitz, M.; Miloud, T.; Kapinsky, M.; Reed, M.R.; Magari, R.; Geissler, E.K.; Hutchinson, J.A.; Vogt, K.; Schlickeiser, S.; Kverneland, A.; et al. Standardization of whole blood immune phenotype monitoring for clinical trials: Panels and methods from the ONE study. Transplant. Res. 2013, 2, 17. [Google Scholar] [CrossRef]
- Michalska-Jakubus, M.; Kowal-Bielecka, O.; Smith, V.; Cutolo, M.; Krasowska, D. Plasma endothelial microparticles reflect the extent of capillaroscopic alterations and correlate with the severity of skin involvement in systemic sclerosis. Microvasc. Res. 2017, 110, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, M.; Pietrowska, M.; Matysiak, N.; Mielańczyk, Ł.; Widłak, P. Proteome Profiling of Exosomes Purified from a Small Amount of Human Serum: The Problem of Co-Purified Serum Components. Proteomes 2019, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; Lanuti, P.; Cicalini, I.; Cufaro, M.C.; Ciccocioppo, F.; Ronci, M.; Simeone, P.; Onofrj, M.; van der Pol, E.; Fontana, A.; et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J. Proteom. 2019, 204, 103403. [Google Scholar] [CrossRef] [PubMed]
- Joy, A.P.; Ayre, D.C.; Chute, I.C.; Beauregard, A.-P.; Wajnberg, G.; Ghosh, A.; Lewis, S.M.; Ouellette, R.J.; Barnett, D.A. Proteome profiling of extracellular vesicles captured with the affinity peptide Vn96: Comparison of Laemmli and TRIzol© protein-extraction methods. J. Extracell. Vesicles 2018, 7, 1438727. [Google Scholar] [CrossRef] [PubMed]
- van Ierssel, S.H.; Van Craenenbroeck, E.M.; Conraads, V.M.; Van Tendeloo, V.F.; Vrints, C.J.; Jorens, P.G.; Hoymans, V.Y. Flow cytometric detection of endothelial microparticles (EMP): Effects of centrifugation and storage alter with the phenotype studied. Thromb. Res. 2010, 125, 332–339. [Google Scholar] [CrossRef]
- Libregts, S.F.W.M.; Arkesteijn, G.J.A.; Németh, A.; Hoen, E.N.M.; Wauben, M.H.M. Flow cytometric analysis of extracellular vesicle subsets in plasma: Impact of swarm by particles of non-interest. J. Thromb. Haemost. 2018, 16, 1423–1436. [Google Scholar] [CrossRef]
- de Rond, L.; Coumans, F.A.W.; Nieuwland, R.; van Leeuwen, T.G.; van der Pol, E. Deriving Extracellular Vesicle Size From Scatter Intensities Measured by Flow Cytometry. Curr. Protoc. Cytom. 2018, 86, e43. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; De Matteis, S.; Carloni, S.; Lanuti, P.; Marchisio, M.; Miscia, S.; Rizzello, A.; Napolitano, R.; Agostinelli, C.; et al. Comparative proteomic profiling of Hodgkin lymphoma cell lines. Mol. Biosyst. 2016, 12, 219–232. [Google Scholar] [CrossRef]
- Platonova, N.; Parravicini, C.; Sensi, C.; Paoli, A.; Colombo, M.; Neri, A.; Eberini, I.; Chiaramonte, R. Identification of small molecules uncoupling the Notch: Jagged interaction through an integrated high-throughput screening. PLoS ONE 2017, 12, e0182640. [Google Scholar] [CrossRef]
- Nolan, J.P.; Jones, J.C. Detection of platelet vesicles by flow cytometry. Platelets 2017, 28, 256–262. [Google Scholar] [CrossRef]
- Celia, C.; Ferrati, S.; Bansal, S.; van de Ven, A.L.; Ruozi, B.; Zabre, E.; Hosali, S.; Paolino, D.; Sarpietro, M.G.; Fine, D.; et al. Sustained zero-order release of intact ultra-stable drug-loaded liposomes from an implantable nanochannel delivery system. Adv. Healthc. Mater. 2014, 3, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Paolino, D.; De Angelis, F.; Cilurzo, F.; Celia, C.; Di Marzio, L.; Russo, D.; Tsapis, N.; Fattal, E.; Fresta, M. Aqueous-core PEG-coated PLA nanocapsules for an efficient entrapment of water soluble anticancer drugs and a smart therapeutic response. Eur. J. Pharm. Biopharm. 2015, 89, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Cicalini, I.; Cufaro, M.C.; Agnifili, L.; Mastropasqua, L.; Lanuti, P.; Marchisio, M.; De Laurenzi, V.; Del Boccio, P.; Pieragostino, D. Multi-Omics Approach for Studying Tears in Treatment-Naïve Glaucoma Patients. Int. J. Mol. Sci. 2019, 20, 4029. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistic | Total EVs/μL | Endo EVs/μL | Leuko EVs/μL | PLT EVs/μL |
|---|---|---|---|---|
| No. of observations | 22.00 | 22.00 | 22.00 | 22.00 |
| Minimum | 613.20 | 0.00 | 71.40 | 134.40 |
| Maximum | 15,569.40 | 2695.00 | 2716.00 | 4044.60 |
| Median | 8997.80 | 136.50 | 361.20 | 1590.90 |
| Mean | 8323.57 | 346.63 | 621.06 | 1898.31 |
| Standard deviation (n-1) | 4214.35 | 601.56 | 742.61 | 1182.74 |
| Statistic | % Endo EVs | % Leuko EVs | % PLT EVs |
|---|---|---|---|
| No. of observations | 22 | 22 | 22 |
| Minimum | 0.00 | 1.1 | 4.5 |
| Maximum | 17.5 | 31.4 | 62.0 |
| Median | 1.9 | 4.6 | 22.1 |
| Mean | 3.6 | 7.8 | 24.8 |
| Standard deviation (n-1) | 4.6 | 7.6 | 13.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchisio, M.; Simeone, P.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Pieragostino, D.; Ventrella, A.; Antonini, F.; Del Zotto, G.; Vergara, D.; et al. Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples. Int. J. Mol. Sci. 2021, 22, 48. https://doi.org/10.3390/ijms22010048
Marchisio M, Simeone P, Bologna G, Ercolino E, Pierdomenico L, Pieragostino D, Ventrella A, Antonini F, Del Zotto G, Vergara D, et al. Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples. International Journal of Molecular Sciences. 2021; 22(1):48. https://doi.org/10.3390/ijms22010048
Chicago/Turabian StyleMarchisio, Marco, Pasquale Simeone, Giuseppina Bologna, Eva Ercolino, Laura Pierdomenico, Damiana Pieragostino, Alessia Ventrella, Francesca Antonini, Genny Del Zotto, Daniele Vergara, and et al. 2021. "Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples" International Journal of Molecular Sciences 22, no. 1: 48. https://doi.org/10.3390/ijms22010048
APA StyleMarchisio, M., Simeone, P., Bologna, G., Ercolino, E., Pierdomenico, L., Pieragostino, D., Ventrella, A., Antonini, F., Del Zotto, G., Vergara, D., Celia, C., Di Marzio, L., Del Boccio, P., Fontana, A., Bosco, D., Miscia, S., & Lanuti, P. (2021). Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples. International Journal of Molecular Sciences, 22(1), 48. https://doi.org/10.3390/ijms22010048