Adaptation to Endoplasmic Reticulum Stress Enhances Resistance of Oral Cancer Cells to Cisplatin by Up-Regulating Polymerase η and Increasing DNA Repair Efficiency

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results
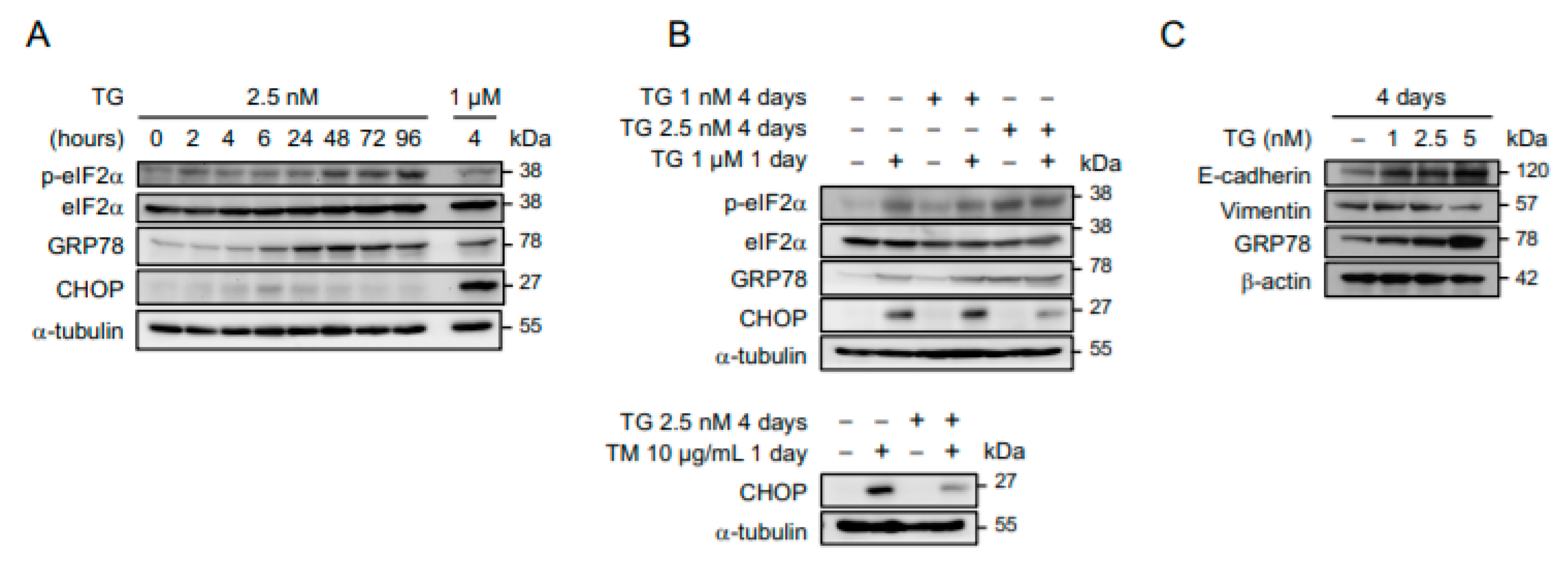
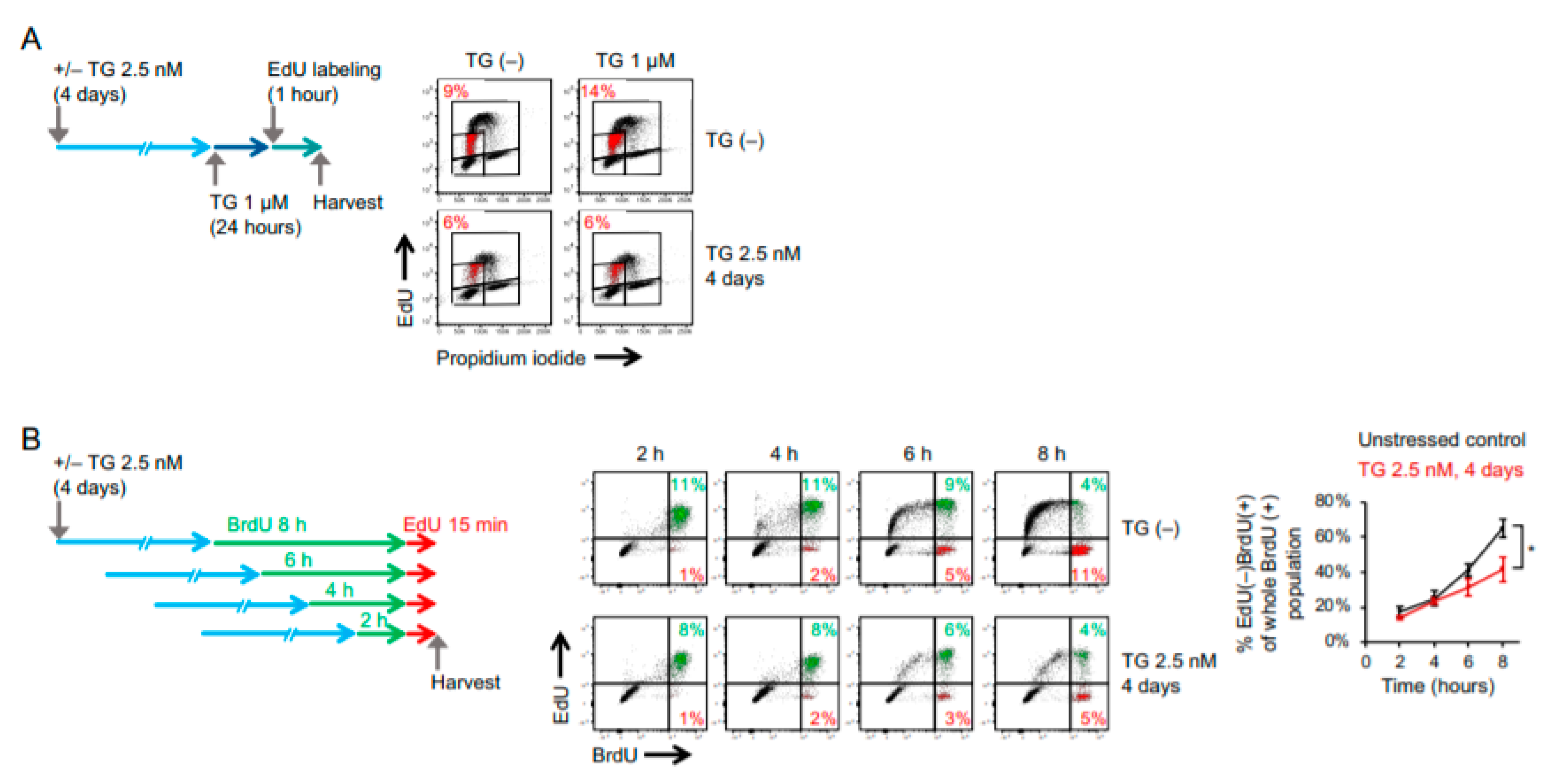
2.1. Cancer Cells Adapt to Persistent ER Stress with Phenotypic Transition
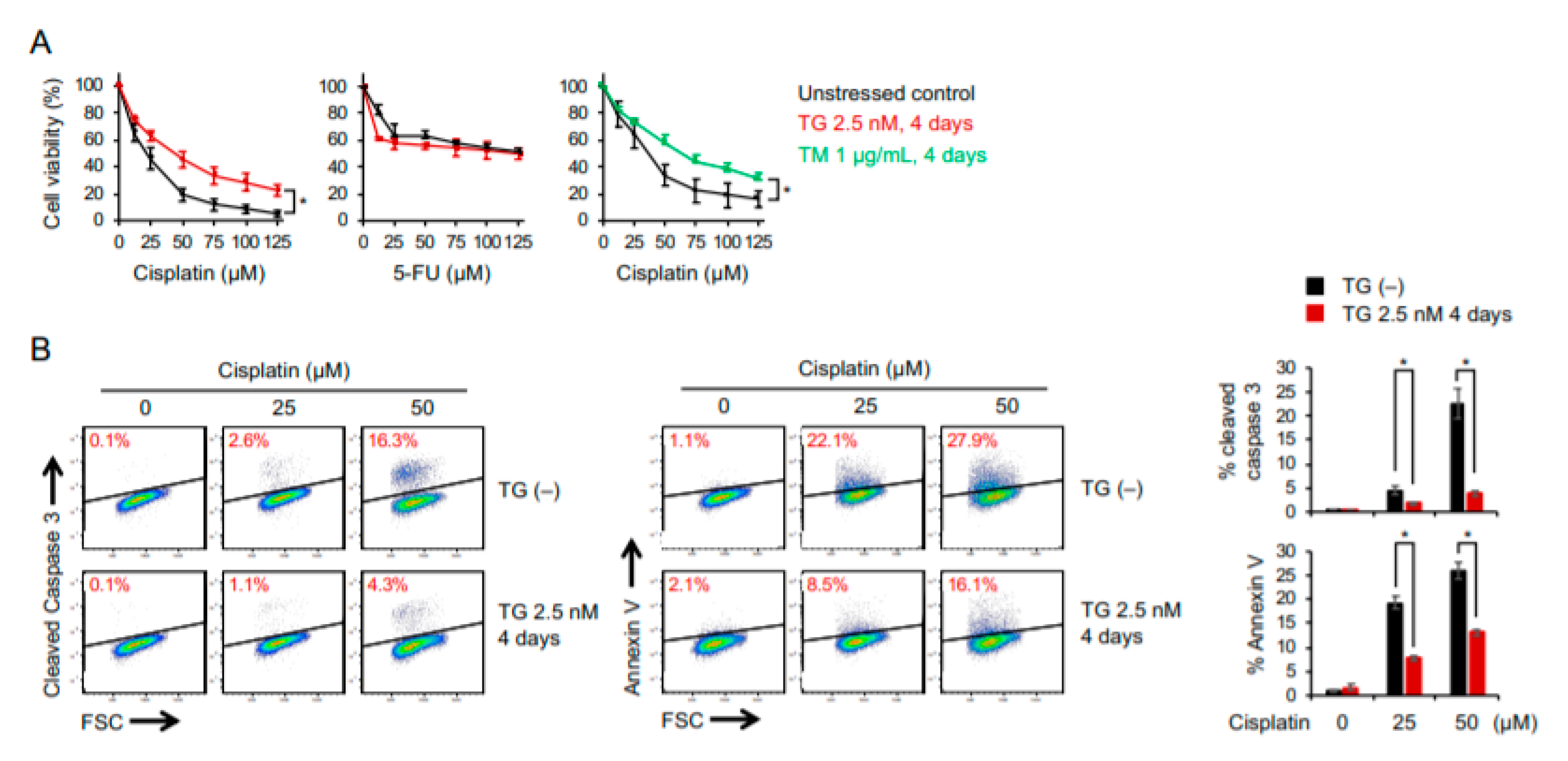
2.2. Cancer Cells Adaptive to ER Stress Are More Resistant to Cisplatin-Induced Cytotoxicity
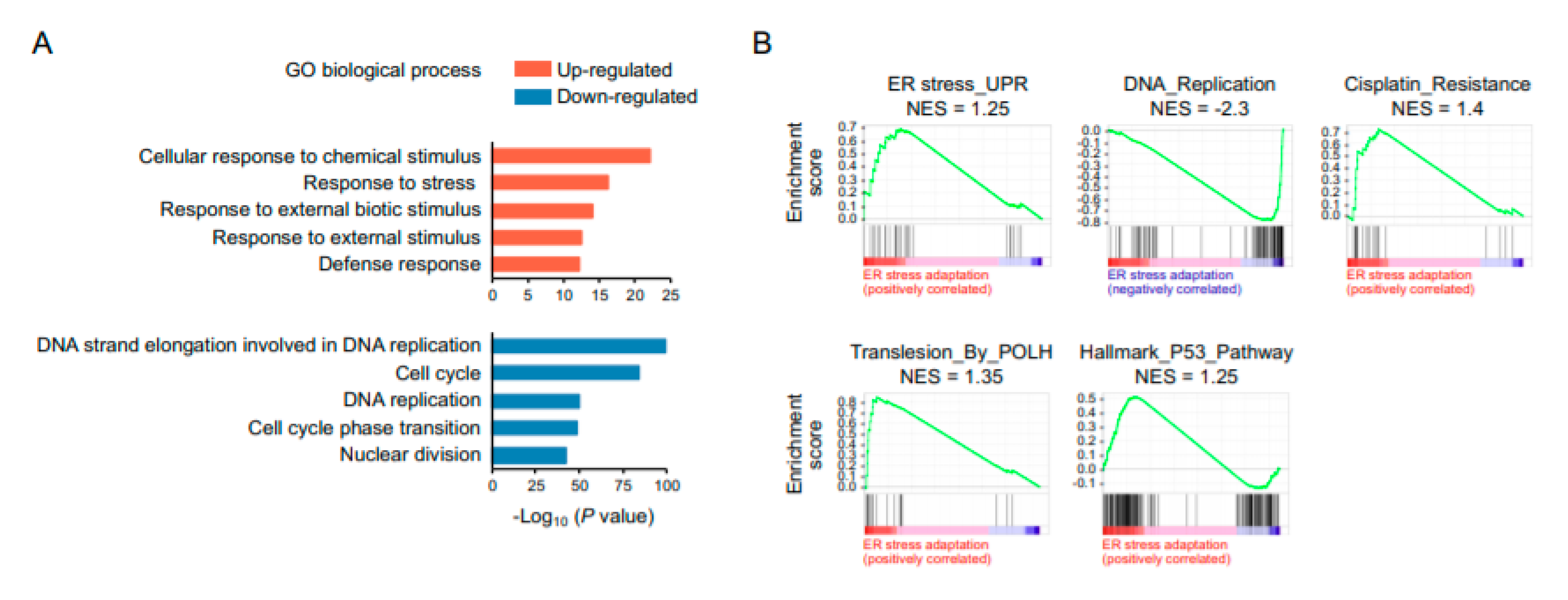
2.3. RNA Sequencing Analysis Reveals a Global Suppression of DNA Replication and Cell-Cycle Progression in ER Stress-Adaptive Cells
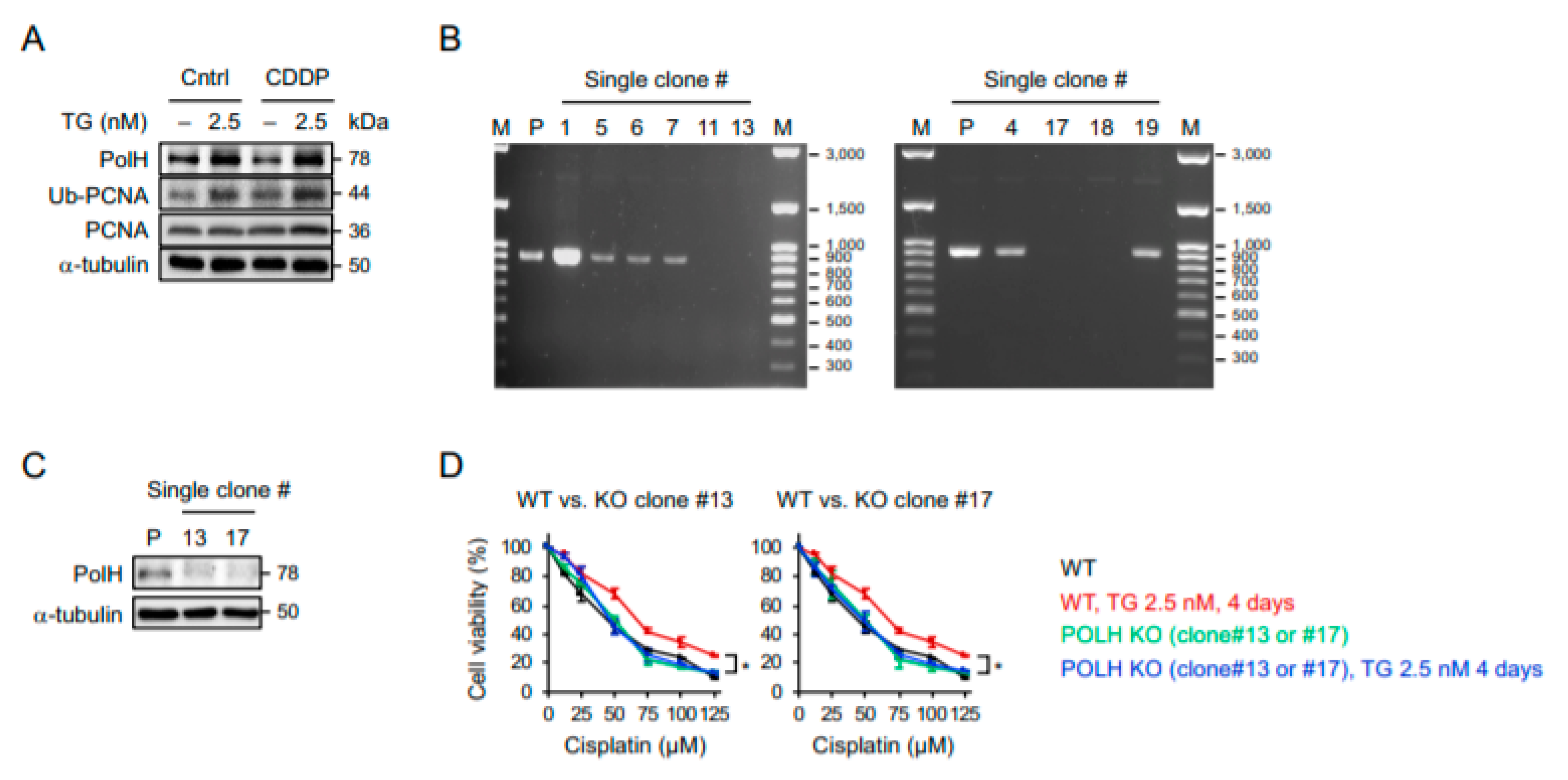
2.4. POLH-Encoded Polymerase η Is Up-Regulated by Prolonged ER Stress and Contributes to Cisplatin Resistance
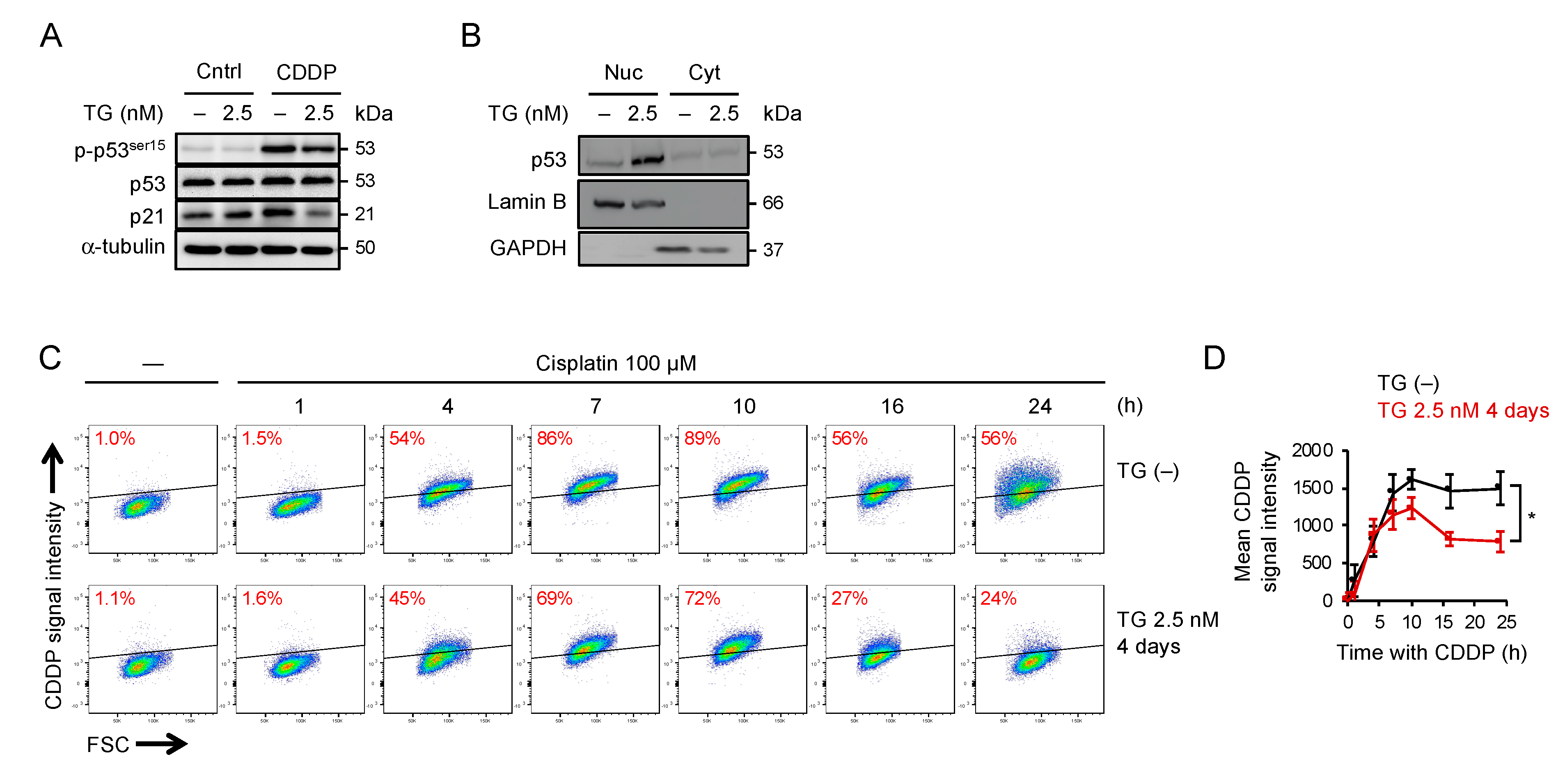
2.5. Adaptation to ER Stress Promotes p53 Nuclear Translocation and Limited the Accumulation of Cisplatin-DNA Lesions
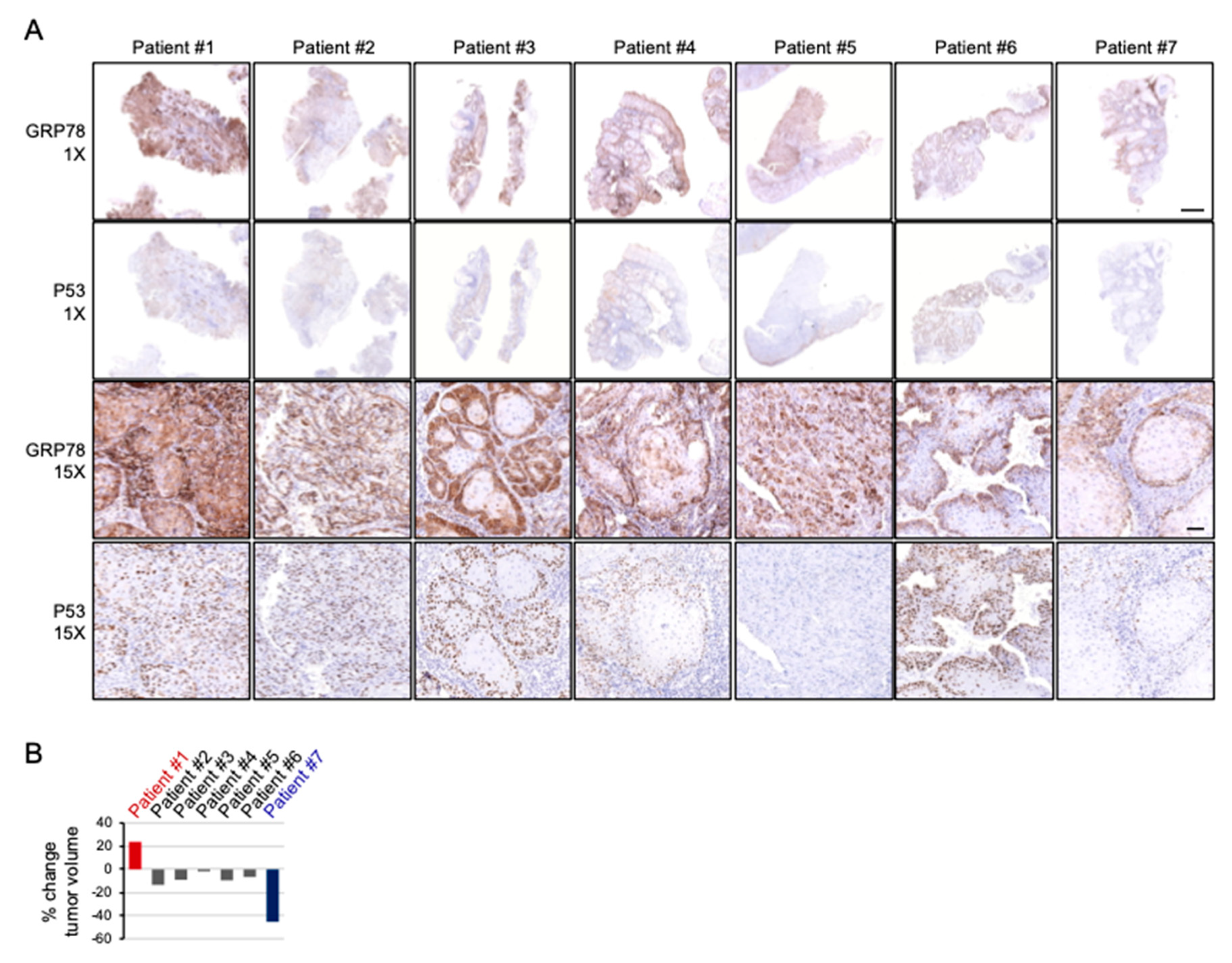
2.6. Cispltain-Based Chemotherapy May Be Less Effective for Patients with Oral Cancers Exhibiting High Levels of ER Stress
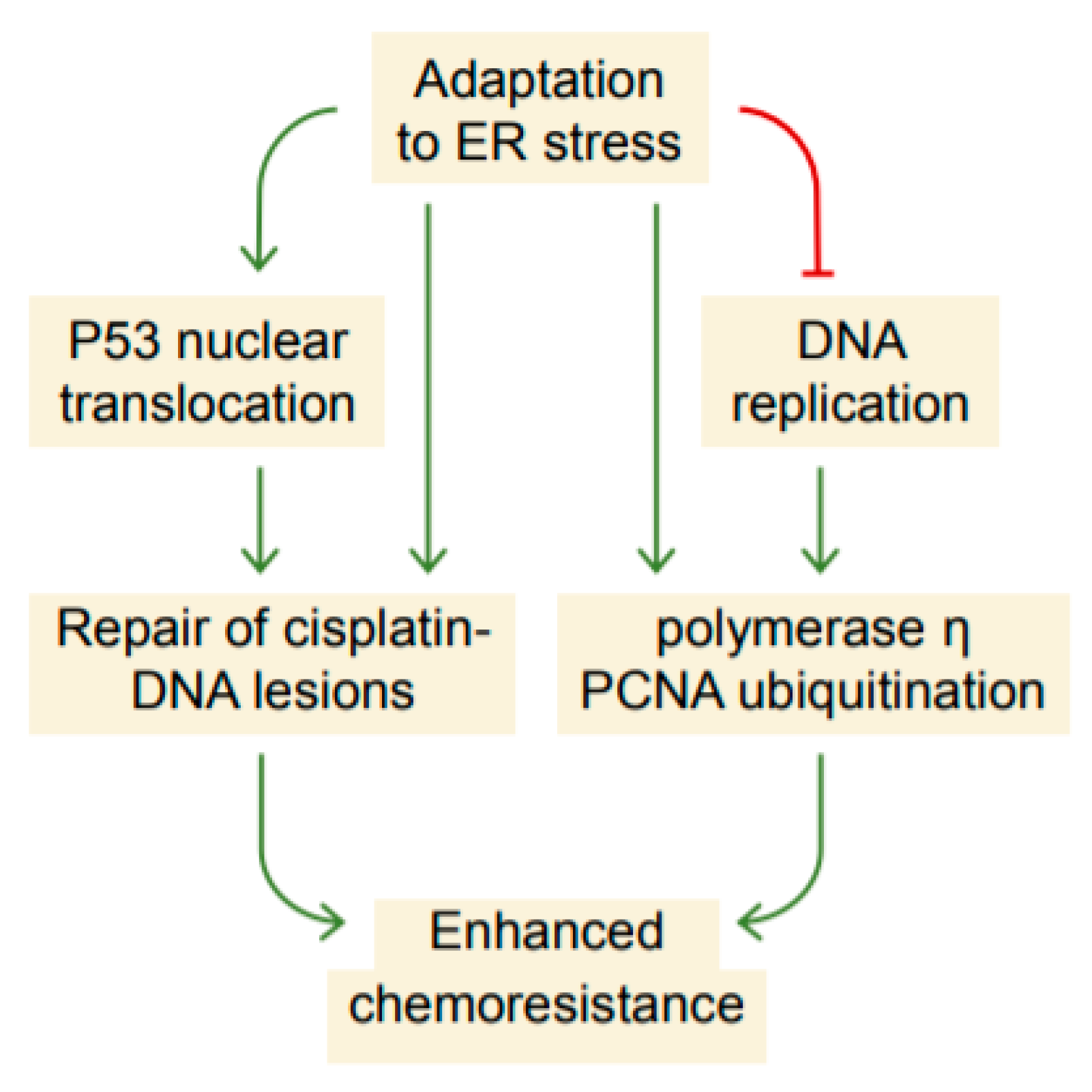
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Patients and Tissue Samples
4.3. Western Blot
4.4. Flow Cytometry Analyses
4.5. WST8 Assay
4.6. RNA Sequencing and Analyses
4.7. CRISPR-Mediated POLH Knockout
4.8. Immunofluorescence
4.9. Genomic DNA Extraction and Targeted Sequencing
4.10. TP53 Knockdown
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic Reticulum Stress: Cell Life and Death Decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. Er Stress and Distinct Outputs of the Ire1alpha Rnase Control Proliferation and Senescence in Response to Oncogenic Ras. Proc. Natl. Acad. Sci. USA 2017, 114, 9900–9905. [Google Scholar] [CrossRef] [PubMed]
- Bright, M.D.; Clarke, P.A.; Workman, P.; Davies, F.E. Oncogenic Rac1 and Nras Drive Resistance to Endoplasmic Reticulum Stress through Mek/Erk Signalling. Cell Signal 2018, 44, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. The Role of the Unfolded Protein Response in Tumour Development: Friend or Foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating Transcription Factor 4 Is Translationally Regulated by Hypoxic Stress. Mol. Cell Biol. 2004, 24, 7469–7482. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of Protein Synthesis by Hypoxia Via Activation of the Endoplasmic Reticulum Kinase Perk and Phosphorylation of the Translation Initiation Factor Eif2alpha. Mol. Cell Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. Ire1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the Xbp-1 Mrna. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. Xbp1 Mrna Is Induced by Atf6 and Spliced by Ire1 in Response to Er Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor Atf6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Rao, R.V.; Peel, A.; Logvinova, A.; del Rio, G.; Hermel, E.; Yokota, T.; Goldsmith, P.C.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling Endoplasmic Reticulum Stress to the Cell Death Program: Role of the Er Chaperone Grp78. FEBS Lett. 2002, 514, 122–128. [Google Scholar] [CrossRef]
- Jamora, C.; Dennert, G.; Lee, A.S. Inhibition of Tumor Progression by Suppression of Stress Protein Grp78/Bip Induction in Fibrosarcoma B/C10me. Proc. Natl. Acad. Sci. USA 1996, 93, 7690–7694. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian Cancer: Strategies for Overcoming Resistance to Chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. Er Stress-Regulated Translation Increases Tolerance to Extreme Hypoxia and Promotes Tumor Growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wei, Q.; Wang, X.; Tang, S.; Liu, H.; Zhang, F.; Mohammed, M.K.; Huang, J.; Guo, D.; Lu, M.; et al. Transition to Resistance: An Unexpected Role of the Emt in Cancer Chemoresistance. Genes. Dis. 2016, 3, 3–6. [Google Scholar] [CrossRef]
- Rossari, F.; Zucchinetti, C.; Buda, G.; Orciuolo, E. Tumor Dormancy as an Alternative Step in the Development of Chemoresistance and Metastasis—Clinical Implications. Cell Oncol. 2020, 43, 155–176. [Google Scholar] [CrossRef]
- Friedberg, E.C. Suffering in Silence: The Tolerance of DNA Damage. Nat. Rev. Mol. Cell Biol. 2005, 6, 943–953. [Google Scholar] [CrossRef]
- Yang, W. Damage Repair DNA Polymerases, Y. Curr. Opin. Struct. Biol. 2003, 13, 23–30. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Fischhaber, P.L.; Kisker, C. Error-Prone DNA Polymerases: Novel Structures and the Benefits of Infidelity. Cell 2001, 107, 9–12. [Google Scholar] [CrossRef]
- Chen, Y.W.; Cleaver, J.E.; Hanaoka, F.; Chang, C.F.; Chou, K.M. A Novel Role of DNA Polymerase Eta in Modulating Cellular Sensitivity to Chemotherapeutic Agents. Mol. Cancer Res. 2006, 4, 257–265. [Google Scholar] [CrossRef]
- Bassett, E.; Vaisman, A.; Havener, J.M.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Efficiency of Extension of Mismatched Primer Termini across from Cisplatin and Oxaliplatin Adducts by Human DNA Polymerases Beta and Eta in Vitro. Biochemistry 2003, 42, 14197–14206. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Efficient Translesion Replication Past Oxaliplatin and Cisplatin Gpg Adducts by Human DNA Polymerase Eta. Biochemistry 2000, 39, 4575–4580. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.D.; Araujo, S.J.; Ariza, R.R.; Batty, D.P.; Biggerstaff, M.; Evans, E.; Gaillard, P.H.; Gunz, D.; Koberle, B.; Kuraoka, I.; et al. DNA Damage Recognition and Nucleotide Excision Repair in Mammalian Cells. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. DNA Excision Repair. Annu. Rev. Biochem. 1996, 65, 43–81. [Google Scholar] [CrossRef]
- Hanawalt, P.C. Subpathways of Nucleotide Excision Repair and Their Regulation. Oncogene 2002, 21, 8949–8956. [Google Scholar] [CrossRef]
- Wellinger, R.E.; Thoma, F. Nucleosome Structure and Positioning Modulate Nucleotide Excision Repair in the Non-Transcribed Strand of an Active Gene. EMBO J. 1997, 16, 5046–5056. [Google Scholar] [CrossRef]
- Liu, Y.; Reeves, D.; Kropachev, K.; Cai, Y.; Ding, S.; Kolbanovskiy, M.; Kolbanovskiy, A.; Bolton, J.L.; Broyde, S.; Van Houten, B.; et al. Probing for DNA Damage with Beta-Hairpins: Similarities in Incision Efficiencies of Bulky DNA Adducts by Prokaryotic and Human Nucleotide Excision Repair Systems in Vitro. DNA Repair 2011, 10, 684–696. [Google Scholar] [CrossRef]
- Lee, J.W.; Ratnakumar, K.; Hung, K.F.; Rokunohe, D.; Kawasumi, M. Deciphering UV-Induced DNA Damage Responses to Prevent and Treat Skin Cancer. Photochem. Photobiol. 2020, 96, 478–499. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. Cytoplasmic Functions of the Tumour Suppressor P53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef]
- O’Brate, A.; Giannakakou, P. The Importance of P53 Location: Nuclear or Cytoplasmic Zip Code? Drug Resist. Updates 2003, 6, 313–322. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of P53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. P53 Has a Direct Apoptogenic Role at the Mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo, R.F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. P53 at the Endoplasmic Reticulum Regulates Apoptosis in a Ca2+-Dependent Manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.K.; Francisco, G.; Soltys, D.T.; Rocha, C.R.; Quinet, A.; Vessoni, A.T.; Castro, L.P.; David, T.I.; Bustos, S.O.; Strauss, B.E.; et al. Predominant Role of DNA Polymerase Eta and P53-Dependent Translesion Synthesis in the Survival of Ultraviolet-Irradiated Human Cells. Nucleic Acids Res. 2017, 45, 1270–1280. [Google Scholar] [CrossRef]
- Qu, L.; Huang, S.; Baltzis, D.; Rivas-Estilla, A.M.; Pluquet, O.; Hatzoglou, M.; Koumenis, C.; Taya, Y.; Yoshimura, A.; Koromilas, A.E. Endoplasmic Reticulum Stress Induces P53 Cytoplasmic Localization and Prevents P53-Dependent Apoptosis by a Pathway Involving Glycogen Synthase Kinase-3beta. Genes. Dev. 2004, 18, 261–277. [Google Scholar] [CrossRef]
- Lin, W.C.; Chuang, Y.C.; Chang, Y.S.; Lai, M.D.; Teng, Y.N.; Su, I.J.; Wang, C.C.; Lee, K.H.; Hung, J.H. Endoplasmic Reticulum Stress Stimulates P53 Expression through Nf-Kappab Activation. PLoS ONE 2012, 7, e39120. [Google Scholar]
- Chu, Y.H.; Sibrian-Vazquez, M.; Escobedo, J.O.; Phillips, A.R.; Dickey, D.T.; Wang, Q.; Ralle, M.; Steyger, P.S.; Strongin, R.M. Systemic Delivery and Biodistribution of Cisplatin in Vivo. Mol. Pharm. 2016, 13, 2677–2682. [Google Scholar] [CrossRef]
- Tamura, R.E.; de Vasconcellos, J.F.; Sarkar, D.; Libermann, T.A.; Fisher, P.B.; Zerbini, L.F. Gadd45 Proteins: Central Players in Tumorigenesis. Curr. Mol. Med. 2012, 12, 634–651. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of Misfolded Proteins Prevents Er-Derived Oxidative Stress and Cell Death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic Reticulum Stress Signaling and Chemotherapy Resistance in Solid Cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. Perk Induces Resistance to Cell Death Elicited by Endoplasmic Reticulum Stress and Chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yu, X.; Yuan, M.; Lv, W.; Feng, T.; Bai, R.; Zhong, H. Activation of the Perk-Atf4 Pathway Promotes Chemo-Resistance in Colon Cancer Cells. Sci. Rep. 2019, 9, 3210. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a Novel Perk Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef]
- Visioli, F.; Wang, Y.; Alam, G.N.; Ning, Y.; Rados, P.V.; Nor, J.E.; Polverini, P.J. Glucose-Regulated Protein 78 (Grp78) Confers Chemoresistance to Tumor Endothelial Cells under Acidic Stress. PLoS ONE 2014, 9, e101053. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of Endoplasmic Reticulum Stress-Induced Apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. Perk/Eif2alpha Signaling Protects Therapy Resistant Hypoxic Cells through Induction of Glutathione Synthesis and Protection against Ros. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef]
- Cook, K.L.; Clarke, P.A.; Parmar, J.; Hu, R.; Schwartz-Roberts, J.L.; Abu-Asab, M.; Warri, A.; Baumann, W.T.; Clarke, R. Knockdown of Estrogen Receptor-Alpha Induces Autophagy and Inhibits Antiestrogen-Mediated Unfolded Protein Response Activation, Promoting Ros-Induced Breast Cancer Cell Death. FASEB J. 2014, 28, 3891–3905. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of Ire1 Rnase Activity Modulates the Tumor Cell Secretome and Enhances Response to Chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, K.; Shao, Y.; Feng, X.; Ji, Z.; Chang, B.; Wang, Y.; Xu, L.; Yang, G. Id1 Confers Cancer Cell Chemoresistance through Stat3/Atf6-Mediated Induction of Autophagy. Cell Death Dis. 2020, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Lu, M.K.; Guan, L.K. The Tsc1 and Tsc2 Tumor Suppressors Are Required for Proper Er Stress Response and Protect Cells from Er Stress-Induced Apoptosis. Cell Death Differ. 2011, 18, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Schewe, D.M.; Aguirre-Ghiso, J.A. Atf6alpha-Rheb-Mtor Signaling Promotes Survival of Dormant Tumor Cells in Vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Fu, Y.; Li, J.; Lee, A.S. Grp78/Bip Inhibits Endoplasmic Reticulum Bik and Protects Human Breast Cancer Cells against Estrogen Starvation-Induced Apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic Reticulum Chaperone Protein Grp78 Protects Cells from Apoptosis Induced by Topoisomerase Inhibitors: Role of Atp Binding Site in Suppression of Caspase-7 Activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. Gli2 Inhibition Abrogates Human Leukemia Stem Cell Dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef]
- Baron, B.; Kitagawa, T. Nakamura, K.; Kuramitsu, Y. Isolation of a Growth Factor Stress-Induced Pancreatic Cancer Sub-Population: Insight into Changes Due to Micro-Environment. Cancer Genom. Proteom. 2015, 12, 49–55. [Google Scholar]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of Disseminated Cancer Cell Dormancy: An Awakening Field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. Perk Mediates Cell-Cycle Exit during the Mammalian Unfolded Protein Response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef]
- Zhao, Y.; Biertumpfel, C.; Gregory, M.T.; Hua, Y.J.; Hanaoka, F.; Yang, W. Structural Basis of Human DNA Polymerase Eta-Mediated Chemoresistance to Cisplatin. Proc. Natl. Acad. Sci. USA 2012, 109, 7269–7274. [Google Scholar] [CrossRef]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-Polymerase Mechanisms Dictate Error-Free and Error-Prone Translesion DNA Synthesis in Mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Bassett, E.; King, N.M.; Bryant, M.F.; Hector, S.; Pendyala, L. Chaney, S.G.; Cordeiro-Stone, M. The Role of DNA Polymerase Eta in Translesion Synthesis Past Platinum-DNA Adducts in Human Fibroblasts. Cancer Res. 2004, 64, 6469–6475. [Google Scholar] [CrossRef] [PubMed]
- Mamenta, E.L.; Poma, E.E.; Kaufmann, W.K.; Delmastro, D.A.; Grady, H.L.; Chaney, S.G. Enhanced Replicative Bypass of Platinum-DNA Adducts in Cisplatin-Resistant Human Ovarian Carcinoma Cell Lines. Cancer Res. 1994, 54, 3500–3505. [Google Scholar] [PubMed]
- Barnes, R.P.; Tsao, W.C.; Moldovan, G.L.; Eckert, K.A. DNA Polymerase Eta Prevents Tumor Cell-Cycle Arrest and Cell Death During Recovery from Replication Stress. Cancer Res. 2018, 78, 6549–6560. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.E. Enhanced Expression of DNA Polymerase Eta Contributes to Cisplatin Resistance of Ovarian Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, P.; Novello, S.; Cambieri, A.; Longo, M.; Monica, V.; Iacono, L.; Giaj-Levra, M.; Saviozzi, S.; Volante, M.; Papotti, M.; et al. Polymerase Eta Mrna Expression Predicts Survival of Non-Small Cell Lung Cancer Patients Treated with Platinum-Based Chemotherapy. Clin. Cancer Res. 2009, 15, 1039–1045. [Google Scholar] [CrossRef]
- Nayak, S.; Calvo, J.A.; Cong, K.; Peng, M.; Berthiaume, E.; Jackson, J.; Zaino, A.M.; Vindigni, A.; Hadden, M.K.; Cantor, S.B. Inhibition of the Translesion Synthesis Polymerase Rev1 Exploits Replication Gaps as a Cancer Vulnerability. Sci. Adv. 2020, 6, eaaz7808. [Google Scholar] [CrossRef]
- Gallo, D.; Kim, T.; Szakal, B.; Saayman, X.; Narula, A.; Park, Y.; Branzei, D.; Zhang, Z.; Brown, G.W. Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of Ssdna Gaps on Undamaged Templates. Mol. Cell 2019, 73, 900–914.e9. [Google Scholar] [CrossRef]
- Daigaku, Y.; Etheridge, T.J.; Nakazawa, Y.; Nakayama, M.; Watson, A.T.; Miyabe, I.; Ogi, T.; Osborne, A.M.; Carr, A.M. Pcna Ubiquitylation Ensures Timely Completion of Unperturbed DNA Replication in Fission Yeast. PLoS Genet. 2017, 13, e1006789. [Google Scholar] [CrossRef]
- Bienko, M.; Green, C.M.; Sabbioneda, S.; Crosetto, N.; Matic, I.; Hibbert, R.G.; Begovic, T.; Niimi, A.; Mann, M.; Lehmann, A.R.; et al. Regulation of Translesion Synthesis DNA Polymerase Eta by Monoubiquitination. Mol. Cell 2010, 37, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Belluscio, L.; van Cappellen, W.A.; Kanaar, R.; Essers, J.; Gottifredi, V. DNA Damage Induced Pol Eta Recruitment Takes Place Independently of the Cell Cycle Phase. Cell Cycle 2009, 8, 3340–3348. [Google Scholar] [CrossRef] [PubMed]
- Sertic, S.; Mollica, A.; Campus, I.; Roma, S.; Tumini, E.; Aguilera, A.; Muzi-Falconi, M. Coordinated Activity of Y Family Tls Polymerases and Exo1 Protects Non-S Phase Cells from Uv-Induced Cytotoxic Lesions. Mol. Cell 2018, 70, 34–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Stavridi, E.S.; Halazonetis, T.D. P53 and Stress in the Er. Genes. Dev. 2004, 18, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Dicks, N.; Bohrer, R.C.; Gutierrez, K.; Michalak, M.; Agellon, L.B.; Bordignon, V. Relief of Endoplasmic Reticulum Stress Enhances DNA Damage Repair and Improves Development of Pre-Implantation Embryos. PLoS ONE 2017, 12, e0187717. [Google Scholar] [CrossRef] [PubMed]
- Dufey, E.; Urra, H.; Hetz, C. Er Proteostasis Addiction in Cancer Biology: Novel Concepts. Semin. Cancer Biol. 2015, 33, 40–47. [Google Scholar] [CrossRef]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. Er Stress Suppresses DNA Double-Strand Break Repair and Sensitizes Tumor Cells to Ionizing Radiation by Stimulating Proteasomal Degradation of Rad51. FEBS Lett. 2013, 578, 3348–3353. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. Xbp1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Xia, L.; Paik, A.; Li, J.J. P53 Activation in Chronic Radiation-Treated Breast Cancer Cells: Regulation of Mdm2/P14arf. Cancer Res. 2004, 64, 221–228. [Google Scholar] [CrossRef]
- Lin, S.C.; Liu, C.J.; Chiu, C.P.; Chang, S.M.; Lu, S.Y.; Chen, Y.J. Establishment of Oc3 Oral Carcinoma Cell Line and Identification of Nf-Kappa B Activation Responses to Areca Nut Extract. J. Oral Pathol. Med. 2004, 33, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, S.; Sakata, K.Y.; Liang, S.; Yoshikawa, K.; Iizasa, H.; Tada, M.; Hamada, J.I.; Kashiwazaki, H.; Kitagawa, Y.; Yamazaki, Y. Dominant-Negative P53 Mutant R248q Increases the Motile and Invasive Activities of Oral Squamous Cell Carcinoma Cells. Biomed. Res. 2019, 40, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. Erdj5 Is Required as a Disulfide Reductase for Degradation of Misfolded Proteins in the Er. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. Cellular Metabolic Stress: Considering How Cells Respond to Nutrient Excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-Ros Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. DNA Damage Checkpoints: From Initiation to Recovery or Adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High Speed of Fork Progression Induces DNA Replication Stress and Genomic Instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Yang, C.Y.; Meng, C.L. Regulation of Pg Synthase by Egf and Pdgf in Human Oral, Breast, Stomach, and Fibrosarcoma Cancer Cell Lines. J. Dent. Res. 1994, 73, 1407–1415. [Google Scholar] [CrossRef]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2020; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 November 2019).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. Gencode Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Feng, J.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Shirley, L.X.; Zhang, Y. Gfold: A Generalized Fold Change for Ranking Differentially Expressed Genes from Rna-Seq Data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for Rna-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (Msigdb) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID. | Tumor Site | Tumor Staging | Radiographic Measurement before Treatment | Surgical Specimen Measurement after Treatment | % Change in Tumor Volume | GRP78 Pattern and Intensity | TP53 Exon | TP53 Mutation |
|---|---|---|---|---|---|---|---|---|
| 1 | Buccal | T4aN1M0 | 60 × 35 × 45 mm | 65 × 45 × 40 mm | 23.80% | Strong Diffuse | 4 8 | P72R E271K |
| 2 | Gingival | T4aN0M0 | 59 × 32 × 45 mm | 50 × 35 × 42 mm | −13.49% | Moderate Diffuse | 4 | P72R |
| 3 | Buccal | T3N1M0 | 45 × 22 × 15 mm | 45 × 30 × 10 mm | −9.09% | Strong Peripheral | 4 | P72R |
| 4 | Gingival | T4aN1M0 | 38 × 36 × 20 mm | 43 × 25 × 25 mm | −1.77% | Moderate Peripheral | 4 | P72R |
| 5 | Tongue | T3N0M0 | 36 × 20 × 25 mm | 38 × 30 × 20 mm | −9.52% | Moderate Diffuse | 9 | S315YS |
| 6 | Buccal | T4aN1M0 | 40 × 27 × 11 mm | 36 × 22 × 14 mm | −6.67% | Moderate Peripheral | 7 | M237I |
| 7 | Tongue | T4aN0M0 | 38 × 33 × 27 mm | 30 × 28 × 22 mm | −45.42% | Weak Peripheral | 4 7 | P72R R248Q |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Kawasumi, M.; Lan, T.-Y.; Poon, C.-L.; Lin, Y.-S.; Wu, P.-J.; Chen, Y.-C.; Chen, B.-H.; Wu, C.-H.; Lo, J.-F.; et al. Adaptation to Endoplasmic Reticulum Stress Enhances Resistance of Oral Cancer Cells to Cisplatin by Up-Regulating Polymerase η and Increasing DNA Repair Efficiency. Int. J. Mol. Sci. 2021, 22, 355. https://doi.org/10.3390/ijms22010355
Chen C-Y, Kawasumi M, Lan T-Y, Poon C-L, Lin Y-S, Wu P-J, Chen Y-C, Chen B-H, Wu C-H, Lo J-F, et al. Adaptation to Endoplasmic Reticulum Stress Enhances Resistance of Oral Cancer Cells to Cisplatin by Up-Regulating Polymerase η and Increasing DNA Repair Efficiency. International Journal of Molecular Sciences. 2021; 22(1):355. https://doi.org/10.3390/ijms22010355
Chicago/Turabian StyleChen, Cho-Yi, Masaoki Kawasumi, Tien-Yun Lan, Chi-Lam Poon, Yi-Sian Lin, Pin-Jou Wu, Yao-Chung Chen, Bing-Hong Chen, Cheng-Hsien Wu, Jeng-Fan Lo, and et al. 2021. "Adaptation to Endoplasmic Reticulum Stress Enhances Resistance of Oral Cancer Cells to Cisplatin by Up-Regulating Polymerase η and Increasing DNA Repair Efficiency" International Journal of Molecular Sciences 22, no. 1: 355. https://doi.org/10.3390/ijms22010355
APA StyleChen, C.-Y., Kawasumi, M., Lan, T.-Y., Poon, C.-L., Lin, Y.-S., Wu, P.-J., Chen, Y.-C., Chen, B.-H., Wu, C.-H., Lo, J.-F., Weng, R. R., Sun, Y.-C., & Hung, K.-F. (2021). Adaptation to Endoplasmic Reticulum Stress Enhances Resistance of Oral Cancer Cells to Cisplatin by Up-Regulating Polymerase η and Increasing DNA Repair Efficiency. International Journal of Molecular Sciences, 22(1), 355. https://doi.org/10.3390/ijms22010355