Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy

 , , and
, , and
Abstract
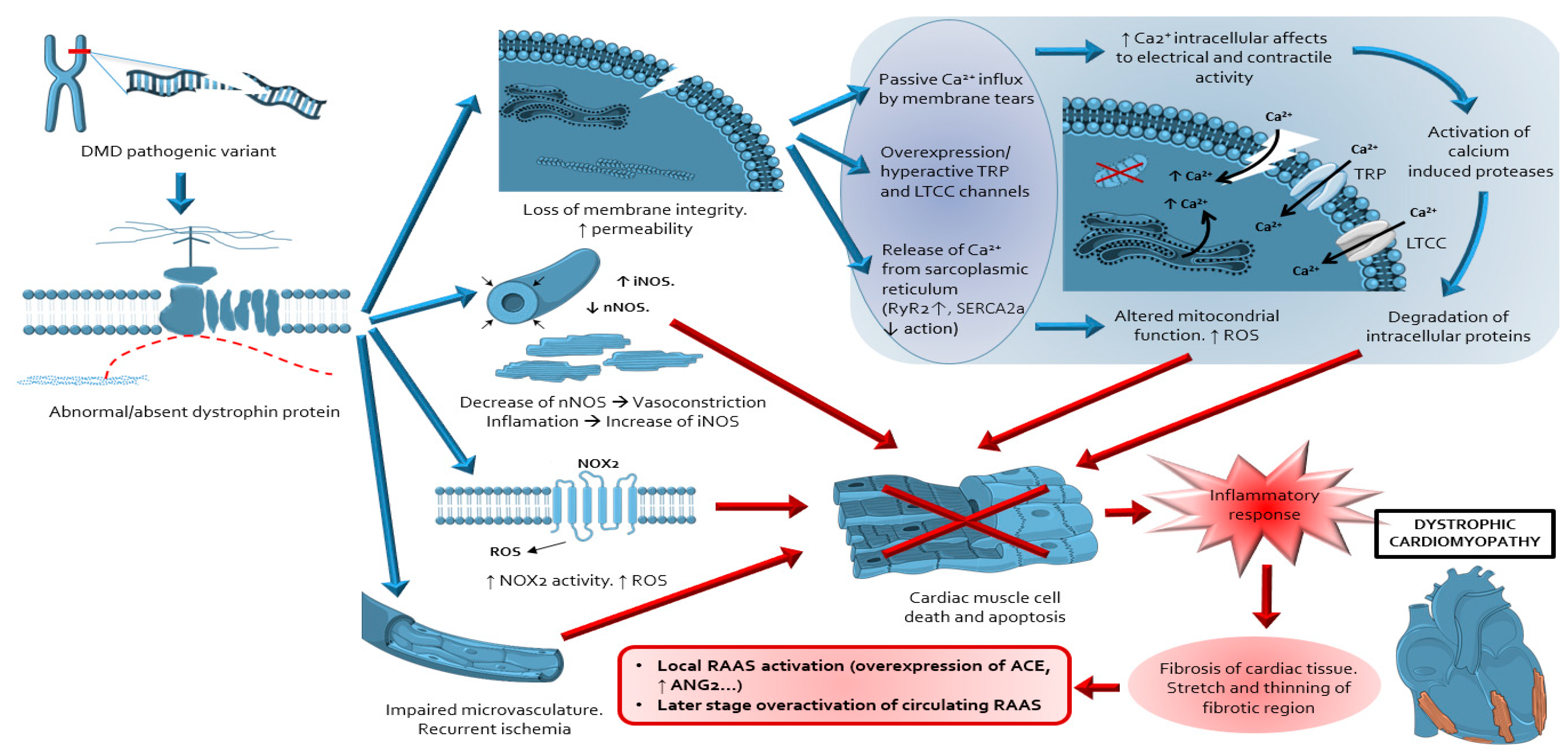
1. Introduction
2. Overview of the Mouse Models Used for the Study of DDC
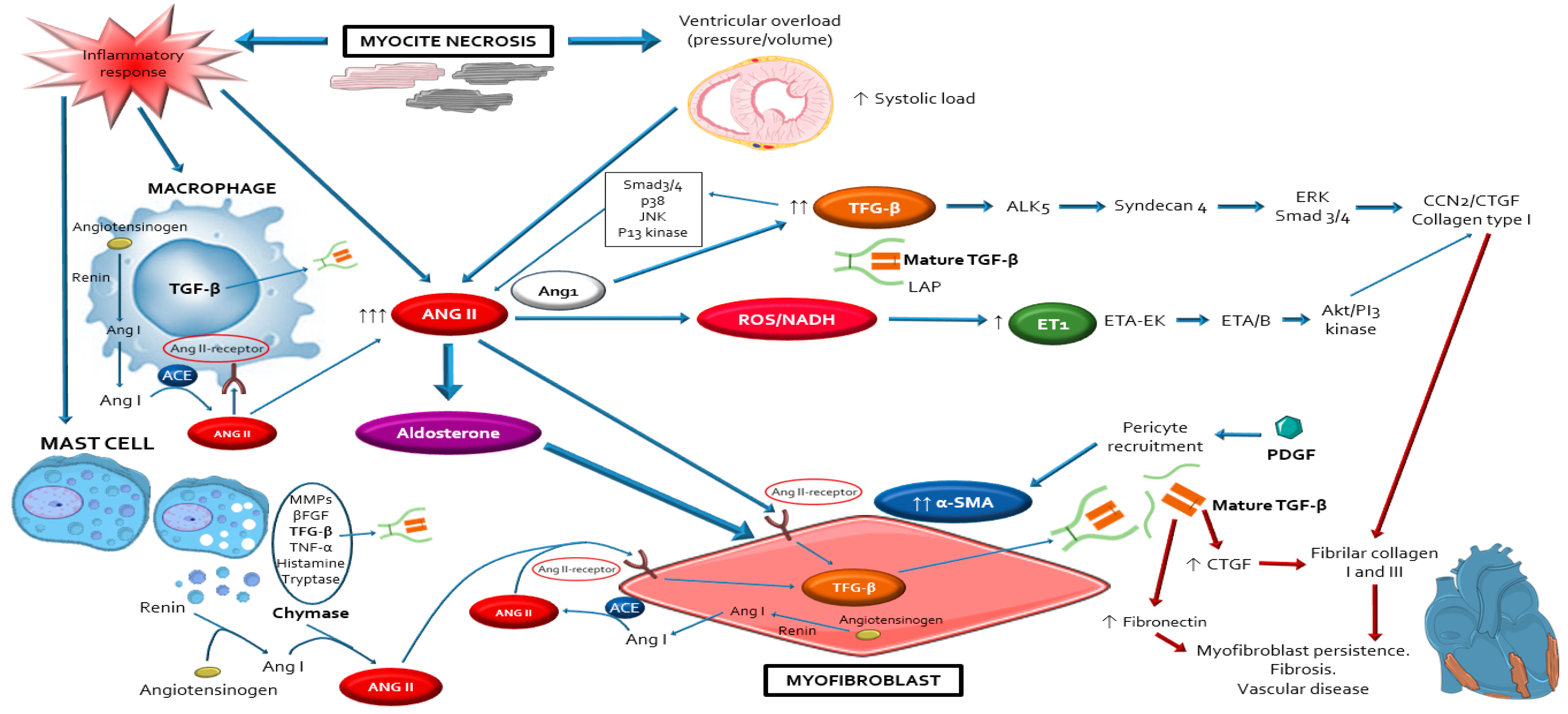
3. Overview of the Fibrotic Process in DDC
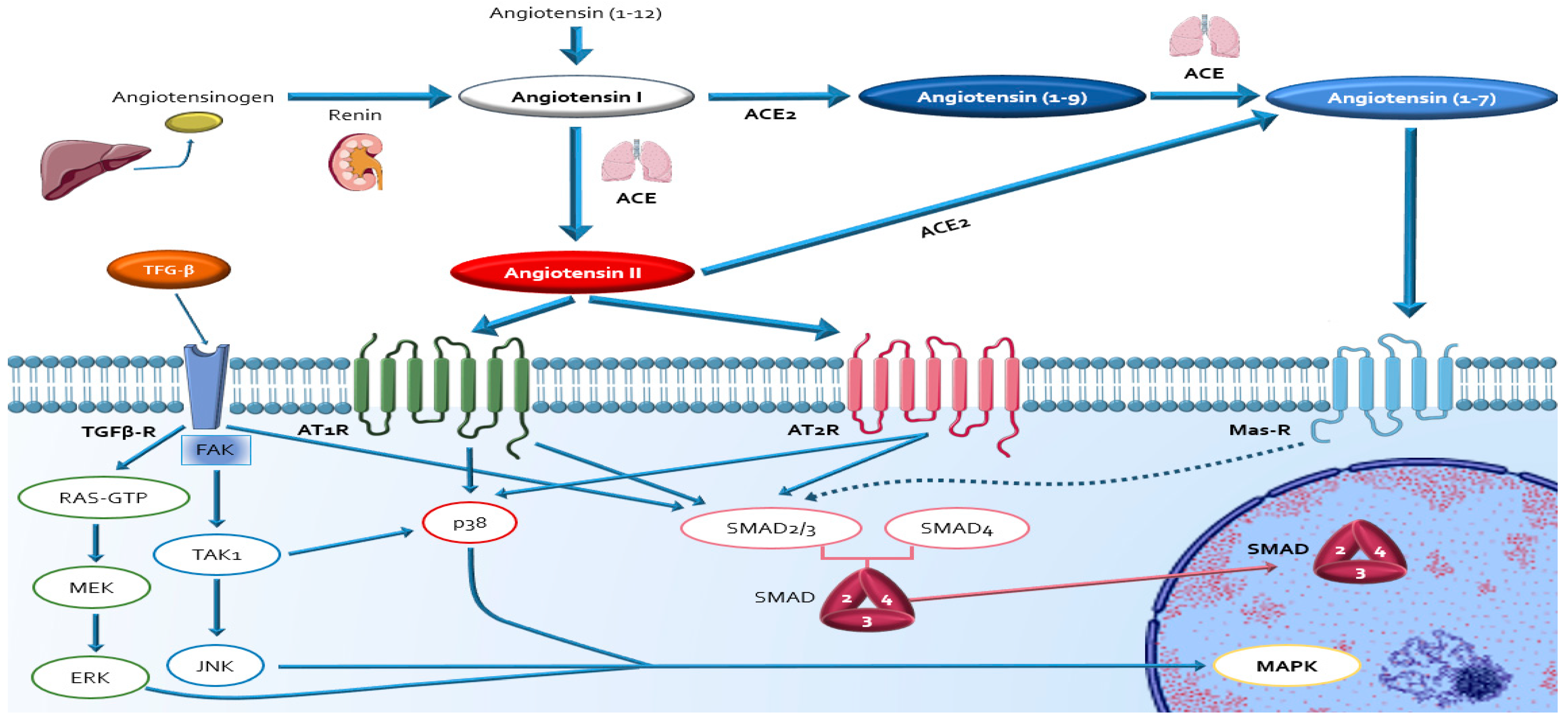
4. Evidence about Sources, Activation and Actions of RAAS in DDC
4.1. Circulating RAAS
4.2. Local Cardiac RAAS
5. Evidence about the Effects of the RAAS Blockade on MF in DDC
5.1. Preclinical Evidence from Mice Models
5.1.1. Single Therapy with ACEI
5.1.2. Single Therapy with ARBs
5.1.3. Single Therapy with MRa
5.1.4. Combined ACEI/ARBs Plus MRa Therapy
5.1.5. Limitations to Translate Preclinical Results to the Clinical Practice in DDC
5.2. Clinical Evidence from Human Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| Akt | Protein kinase B |
| ALK5 | Activin receptor-like kinase-1 |
| Ang I | Angiotensin I |
| ANG II | Angiotensin II |
| ARBs | AT1R blockers |
| AT1R | Angiotensin II receptor type 1 |
| AT2R | Angiotensin II receptor type 2 |
| BMD | Becker muscular dystrophy |
| CCN2/CTGF | connective tissue growth factor |
| CHF | Congestive heart failure |
| CF | Cardiac fibrosis |
| cTn | Cardiac troponin |
| DCM | Dilated cardiomyopathy |
| DDC | Dystrophin-deficient cardiomyopathy |
| DMD | Duchenne muscular dystrophy |
| ERK | Extracellular signal-regulated kinase |
| ET1 | Endothelin-1 |
| ETA | Endothelin receptor A |
| FAK | Focal adhesion kinase |
| JNK | Jun N-Terminal Kinase |
| iNOS | Inducible nitric oxide synthase |
| LTCC | L-type Ca2+ channels |
| LVH | Left ventricular hypertrophy |
| LVSD | Left ventricular systolic dysfunction |
| LVDD | Left ventricular diastolic dysfunction |
| MAPK | Mitogen-activated protein kinase |
| MasR | Mas receptor |
| MEK | MAPK/ERK kinase |
| MF | Myocardial fibrosis |
| MMPs | Matrix metalloproteinases |
| MP | Myopericarditis |
| MRI | Magnetic resonance imaging |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor kappa B |
| nNOS | neuronal nitric oxide synthase |
| NOX2 | NADPH oxidase 2 |
| NP | Natriuretic peptides |
| PDGF | Platelet derived growth factor |
| PI3 | Phosphoinositide 3 |
| ROS | Reactive oxygen species |
| TAK1 | Tumour growth factor β-activating kinase-1 |
| TGF-β | Transforming growth factor-β |
| TGF-βR | Transforming growth factor-β receptor |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TRP | Transient receptor potential |
| RyR2 | Ryanodine receptor 2 |
| SMAD | Mothers against decapentaplegic homolog |
| SERCA2 | Sarcoplasmic/endoplasmic reticulum calcium ATP-ase |
| TFG-β | Transforming growth factor-beta |
| TNF-α | Tumour necrosis factor alpha |
| VA | Ventricular arrhythmias |
| α-SMA | Alpha-smooth muscle actin |
| βFGF | Basic fibroblast growth factor |
References
- Brandsema, J.F.; Darras, B.T. Dystrophinopathies. Semin. Neurol. 2015, 35, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.A.; Mahajan, K. Dystrophinopathies. In Gene Reviews; Stat Pearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [PubMed]
- Le, S.; Yu, M.; Hovan, L.; Zhao, Z.; Ervasti, J.; Yan, J. Dystrophin as a Molecular Shock Absorber. ACS Nano 2018, 12, 12140–12148. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Ebner, J.; Hilber, K. Voltage-Dependent Sarcolemmal Ion Channel Abnormalities in the Dystrophin-Deficient Heart. Int. J. Mol. Sci. 2018, 19, 3296. [Google Scholar] [CrossRef] [PubMed]
- Rubi, L.; Todt, H.; Kubista, H.; Koenig, X.; Hilber, K. Calcium current properties in dystrophin-deficient ventricular cardiomyocytes from aged mdx mice. Physiol. Rep. 2018, 6, e13567. [Google Scholar] [CrossRef]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Shima, A.; Walter, G.A.; Sweeney, H.L. Tadalafil Treatment Delays the Onset of Cardiomyopathy in Dystrophin-Deficient Hearts. J. Am. Heart Assoc. 2016, 5, 5. [Google Scholar] [CrossRef]
- Viola, H.M.; Davies, S.M.; Filipovska, A. Hool LC. L-type Ca(2+) channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H767–H775. [Google Scholar] [CrossRef]
- Johnstone, V.P.; Viola, H.M.; Hool, L.C. Dystrophic Cardiomyopathy-Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies. Genes 2017, 8, 108. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Martin, D.; Schoor, M.C.-V.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Lorin, C.; Vogeli, I.; Niggli, E. Dystrophic cardiomyopathy: Role of TRPV2 channels in stretch-induced cell damage. Cardiovasc. Res. 2015, 106, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; Lopez de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Croall, D.E.; Tidball, J.G. Calpains are activated in necrotic fibers from mdx dystrophic mice. J. Biol. Chem. 1995, 270, 10909–10914. [Google Scholar] [CrossRef] [PubMed]
- Badalamente, M.A.; Stracher, A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve 2000, 23, 106–111. [Google Scholar] [CrossRef]
- Loboda, A.; Dulak, J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: Past, present, and future. Pharmacol. Rep. Pr. 2020, 72, 1227–1263. [Google Scholar] [CrossRef]
- Bia, B.L.; Cassidy, P.J.; Young, M.E.; Rafael, J.A.; Leighton, B.; Davies, K.G.; Radda, G.K.; Clarke, K. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 1999, 31, 1857–1862. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef]
- Dombernowsky, N.W.; Olmestig, J.N.E.; Witting, N.; Kruuse, C. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies-Still a possible treatment modality? Neuromuscul. Disord. NMD 2018, 28, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L.; Ungureanu-Longrois, D.; Simmons, W.W.; Pimental, D.; Malinski, T.A.; Kapturczak, M.; Taha, Z.; Lowenstein, C.J.; Davidoff, A.J.; Kelly, R.A. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J. Biol. Chem. 1994, 269, 27580–27588. [Google Scholar] [PubMed]
- Kyrychenko, V.; Polakova, E.; Janicek, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatric Neurol. 2013, 20, 202–215. [Google Scholar] [CrossRef]
- Buyse, G.M.; Goemans, N.; Hauwe, M.V.D.; Thijs, D.; De Groot, I.J.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. NMD 2011, 21, 396–405. [Google Scholar] [CrossRef]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef]
- Quinlan, J.G.; Hahn, H.S.; Wong, B.L.; Lorenz, J.N.; Wenisch, A.S.; Levin, L.S. Evolution of the mdx mouse cardiomyopathy: Physiological and morphological findings. Neuromuscul. Disord. NMD 2004, 14, 491–496. [Google Scholar] [CrossRef]
- Finsterer, J.; Stollberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef]
- Tandon, A.; Villa, C.R.; Hor, K.N.; Jefferies, J.L.; Gao, Z.; Towbin, J.A.; Wong, B.L.; Mazur, W.; Fleck, R.J.; Sticka, J.J.; et al. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in duchenne muscular dystrophy. J. Am. Heart Assoc. 2015, 4, e001338. [Google Scholar] [CrossRef] [PubMed]
- Adorisio, R.; Mencarelli, E.; Cantarutti, N.; Calvieri, C.; Amato, L.; Cicenia, M.; Silvetti, M.; D’Amico, A.; Grandinetti, M.; Drago, F.; et al. Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies. J. Clin. Med. 2020, 9, 3186. [Google Scholar] [CrossRef] [PubMed]
- Hor, K.N.; Johnston, P.; Kinnett, K.; Mah, M.L.; Stiver, C.; Markham, L.; Cripe, L.H. Progression of Duchenne Cardiomyopathy Presenting with Chest Pain and Troponin Elevation. J. Neuromuscul. Dis. 2017, 4, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Engel, W.K.; Derrer, E.C. Duchenne muscular dystrophy: Functional ischemia reproduces its characteristic lesions. Science 1971, 172, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef]
- Tarbit, E.; Singh, I.; Peart, J.N.; Rose’Meyer, R.B. Biomarkers for the identification of cardiac fibroblast and myofibroblast cells. Heart Fail. Rev. 2019, 24, 1–15. [Google Scholar] [CrossRef]
- Cao, L.; Chen, Y.; Lu, L.; Liu, Y.; Wang, Y.; Fan, J.; Yin, Y. Angiotensin II upregulates fibroblast-myofibroblast transition through Cx43-dependent CaMKII and TGF-beta1 signaling in neonatal rat cardiac fibroblasts. Acta Biochim. Biophys. Sin. 2018, 50, 843–852. [Google Scholar] [CrossRef]
- Power, L.C.; O’Grady, G.L.; Hornung, T.S.; Jefferies, C.; Gusso, S.; Hofman, P.L. Imaging the heart to detect cardiomyopathy in Duchenne muscular dystrophy: A review. Neuromuscul. Disord. Nmd 2018, 28, 717–730. [Google Scholar] [CrossRef]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef]
- Mavrogeni, S.I.; Markousis-Mavrogenis, G.; Papavasiliou, A.; Papadopoulos, G.; Kolovou, G. Cardiac Involvement in Duchenne Muscular Dystrophy and Related Dystrophinopathies. Methods Mol. Biol. 2018, 1687, 31–42. [Google Scholar]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2, respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Haudek, S.B.; Cheng, J.; Du, J.; Wang, Y.; Hermosillo-Rodriguez, J.; Trial, J.; Taffet, G.E.; Entman, M.L. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2010, 49, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Crowley, S.D.; Coffman, T.M. Recent advances involving the renin-angiotensin system. Exp. Cell Res. 2012, 318, 1049–1056. [Google Scholar] [CrossRef]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef]
- Nishimura, T.; Yanagisawa, A.; Sakata, H.; Sakata, K.; Shimoyama, K.; Ishihara, T.; Yoshino, H.; Ishikawa, K. Thallium-201 single photon emission computed tomography (SPECT) in patients with duchenne’s progressive muscular dystrophy: A histopathologic correlation study. Jpn. Circ. J. 2001, 65, 99–105. [Google Scholar] [CrossRef]
- Sanyal, S.K.; Johnson, W.W.; Thapar, M.K.; Pitner, S.E. An ultrastructural basis for electrocardiographic alterations associated with Duchenne’s progressive muscular dystrophy. Circulation 1978, 57, 1122–1129. [Google Scholar] [CrossRef]
- Kamogawa, Y.; Biro, S.; Maeda, M.; Setoguchi, M.; Hirakawa, T.; Yoshida, H.; Tei, C. Dystrophin-deficient myocardium is vulnerable to pressure overload in vivo. Cardiovasc. Res. 2001, 50, 509–515. [Google Scholar] [CrossRef]
- Lefaucheur, J.P.; Sebille, A. Basic fibroblast growth factor promotes in vivo muscle regeneration in murine muscular dystrophy. Neurosci. Lett. 1995, 202, 121–124. [Google Scholar] [CrossRef]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; Van Der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yoshida, K.; Takeda, S.; Dohi, N.; Ikeda, S.-I. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002, 520, 18–24. [Google Scholar] [CrossRef]
- Cohn, R.D.; Durbeej, M.; Moore, S.A.; Coral-Vazquez, R.; Prouty, S.; Campbell, K.P. Prevention of cardiomyopathy in mouse models lacking the smooth muscle sarcoglycan-sarcospan complex. J. Clin. Investig. 2001, 107, R1–R7. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.J.; Barbin, I.C.; Taniguti, A.P.; Oggian, D.S.; Ferretti, R.; Santo Neto, H. Myocardial fibrosis is unaltered by long-term administration of L-arginine in dystrophin deficient mdx mice: A histomorphometric analysis. Acta Biol. Hung. 2010, 61, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef]
- Stempien-Otero, A.; Kim, D.H.; Davis, J. Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell. Cardiol. 2016, 97, 153–161. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Xu, J.; Li, W.; Zhao, D.; Zhao, Y.; Yan, C. Activation of the wnt/beta-Catenin Signaling Pathway in Polymyositis, Dermatomyositis and Duchenne Muscular Dystrophy. J. Clin. Neurol. 2016, 12, 351–360. [Google Scholar] [CrossRef]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef]
- Duerrschmid, C.; Trial, J.; Wang, Y.; Entman, M.L.; Haudek, S.B. Tumor necrosis factor: A mechanistic link between angiotensin-II-induced cardiac inflammation and fibrosis. Circ. Heart Fail. 2015, 8, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin beta1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar]
- Wang, X.; Wang, H.X.; Li, Y.L.; Zhang, C.C.; Zhou, C.Y.; Wang, L.; Xia, Y.L.; Du, J.; Li, H.H. MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension 2015, 66, 776–785. [Google Scholar] [CrossRef]
- Chen, C.; Du, J.; Feng, W.; Song, Y.; Lu, Z.; Xu, M.; Li, Z.; Zhang, Y. beta-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br. J. Pharmacol. 2012, 166, 676–688. [Google Scholar] [CrossRef]
- Liu, N.; Xing, R.; Yang, C.; Tian, A.; Lv, Z.; Sun, N.; Gao, X.; Zhang, Y.; Li, Z. HIP-55/DBNL-dependent regulation of adrenergic receptor mediates the ERK1/2 proliferative pathway. Mol. Biosyst. 2014, 10, 1932–1939. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Schroer, A.K.; Merryman, W.D. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J. Cell Sci. 2015, 128, 1865–1875. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Investig. 2013, 123, 1096–1108. [Google Scholar] [CrossRef]
- Kural, M.H.; Billiar, K.L. Myofibroblast persistence with real-time changes in boundary stiffness. Acta Biomater. 2016, 32, 223–230. [Google Scholar] [CrossRef]
- Li, L.; Fan, D.; Wang, C.; Wang, J.Y.; Cui, X.B.; Wu, D.; Zhou, Y.; Wu, L.L. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-beta1 pathways in cardiac fibroblasts. Cardiovasc. Res. 2011, 91, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, L.; Xie, J.; Li, R.; Li, G.N.; Chen, Q.H.; Zhang, X.L.; Kang, L.N.; Xu, B. Periostin expression induced by oxidative stress contributes to myocardial fibrosis in a rat model of high salt-induced hypertension. Mol. Med. Rep. 2016, 14, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Cody, R.J.; Graf, K.; Goetze, S.; Kawano, Y.; Schnee, J.; Law, R.E.; Hsueh, W.A. Angiotensin II enhances integrin and alpha-actinin expression in adult rat cardiac fibroblasts. Hypertension 2000, 35, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Megeney, L.A.; Kablar, B.; Perry, R.L.; Ying, C.; May, L.; Rudnicki, M.A. Severe cardiomyopathy in mice lacking dystrophin and MyoD. Proc. Natl. Acad. Sci. USA 1999, 96, 220–225. [Google Scholar] [CrossRef]
- Santos, R.A.; Ferreira, A.J.; Simoes, E.S.A.C. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp. Physiol. 2008, 93, 519–527. [Google Scholar] [CrossRef]
- Sun, G.; Haginoya, K.; Dai, H.; Chiba, Y.; Uematsu, M.; Hino-Fukuyo, N.; Onuma, A.; Iinuma, K.; Tsuchiya, S. Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J. Neurol. Sci. 2009, 280, 40–48. [Google Scholar] [CrossRef]
- Zamai, L. The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells 2020, 9, 1704. [Google Scholar] [CrossRef]
- Dinh, D.T.; Frauman, A.G.; Johnston, C.I.; Fabiani, M.E. Angiotensin receptors: Distribution, signalling and function. Clin. Sci. 2001, 100, 481–492. [Google Scholar] [CrossRef]
- Dostal, D.E.; Baker, K.M. The cardiac renin-angiotensin system: Conceptual, or a regulator of cardiac function? Circ. Res. 1999, 85, 643–650. [Google Scholar] [CrossRef]
- Bader, M. Molecular interactions of vasoactive systems in cardiovascular damage. J. Cardiovasc. Pharmacol. 2001, 38 (Suppl. 2), S7–S9. [Google Scholar] [CrossRef]
- Catena, C.; Colussi, G.; Brosolo, G.; Novello, M.; Sechi, L.A. Aldosterone and Left Ventricular Remodeling. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2015, 47, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.S.; Unal, H.; Kemp, J.R.; Tirupula, K.C.; Eguchi, S.; Vanderheyden, P.M.; Thomas, W.G. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharmacol. Rev. 2015, 67, 754–819. [Google Scholar] [CrossRef] [PubMed]
- Serneri, G.G.; Boddi, M.; Cecioni, I.; Vanni, S.; Coppo, M.; Papa, M.L.; Bandinelli, B.; Bertolozzi, I.; Polidori, G.; Toscano, T.; et al. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function. Circ. Res. 2001, 88, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H.; van Kats, J.P.; Admiraal, P.J.; Derkx, F.H.; Lamers, J.M.; Verdouw, P.D.; Saxena, P.R.; Schalekamp, M.A. Cardiac renin and angiotensins. Uptake from plasma versus in situ synthesis. Hypertension 1994, 24, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.C.; Mackins, C.J.; Seyedi, N.; Levi, R.; Silver, R.B. Coupling of angiotensin II AT1 receptors to neuronal NHE activity and carrier-mediated norepinephrine release in myocardial ischemia. Am. J. Physiol. Circ. Physiol. 2004, 286, H1448–H1454. [Google Scholar] [CrossRef] [PubMed]
- Seyedi, N.; Win, T.; Lander, H.M.; Levi, R. Bradykinin B2-receptor activation augments norepinephrine exocytosis from cardiac sympathetic nerve endings. Mediation by autocrine/paracrine mechanisms. Circ. Res. 1997, 81, 774–784. [Google Scholar] [CrossRef]
- Sabharwal, R.; Chapleau, M.W. Autonomic, locomotor and cardiac abnormalities in a mouse model of muscular dystrophy: Targeting the renin-angiotensin system. Exp. Physiol. 2014, 99, 627–631. [Google Scholar] [CrossRef]
- Sabharwal, R.; Weiss, R.M.; Zimmerman, K.; Domenig, O.; Cicha, M.Z.; Chapleau, M.W. Angiotensin-dependent autonomic dysregulation precedes dilated cardiomyopathy in a mouse model of muscular dystrophy. Exp. Physiol. 2015, 100, 776–795. [Google Scholar] [CrossRef]
- Sabharwal, R.; Cicha, M.Z.; Sinisterra, R.D.; De Sousa, F.B.; Santos, R.A.; Chapleau, M.W. Chronic oral administration of Ang-(1-7) improves skeletal muscle, autonomic and locomotor phenotypes in muscular dystrophy. Clin. Sci. 2014, 127, 101–109. [Google Scholar] [CrossRef]
- Nakamura, A.; Harrod, G.V.; Davies, K.E. Activation of calcineurin and stress activated protein kinase/p38-mitogen activated protein kinase in hearts of utrophin-dystrophin knockout mice. Neuromuscul. Disord. Nmd 2001, 11, 251–259. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Chiyo, T.; Nishiyama, A.; Okada, H.; Takeda, S.; Okada, T. Dystrophic mdx mice develop severe cardiac and respiratory dysfunction following genetic ablation of the anti-inflammatory cytokine IL-10. Hum. Mol. Genet. 2014, 23, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Jordan, M.C.; Gotoh, T.; Grody, W.W.; Roos, K.P.; Tidball, J.G. Arginine metabolism by macrophages promotes cardiac and muscle fibrosis in mdx muscular dystrophy. PLoS ONE 2010, 5, e10763. [Google Scholar] [CrossRef] [PubMed]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Hirono, Y.; Yoshimoto, T.; Suzuki, N.; Sugiyama, T.; Sakurada, M.; Takai, S.; Kobayashi, N.; Shichiri, M.; Hirata, Y. Angiotensin II receptor type 1-mediated vascular oxidative stress and proinflammatory gene expression in aldosterone-induced hypertension: The possible role of local renin-angiotensin system. Endocrinology 2007, 148, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- De Mello, W.C.; Frohlich, E.D. Clinical perspectives and fundamental aspects of local cardiovascular and renal Renin-Angiotensin systems. Front. Endocrinol. 2014, 5, 16. [Google Scholar] [CrossRef]
- Uehara, Y.; Miura, S.; Yahiro, E.; Saku, K. Non-ACE pathway-induced angiotensin II production. Curr. Pharm. Des. 2013, 19, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Ahmad, S.; Varagic, J.; Cheng, C.P.; Groban, L.; Wang, H.; Collawn, J.F.; Dell Italia, L.J. Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. Am. J. Physiol. Circ. Physiol. 2016, 311, H404–H414. [Google Scholar] [CrossRef]
- Lowe, J.; Kolkhof, P.; Haupt, M.J.; Peczkowski, K.K.; Rastogi, N.; Hauck, J.S.; Kadakia, F.K.; Zins, J.G.; Ciccone, P.C.; Smart, S.; et al. Mineralocorticoid receptor antagonism by finerenone is sufficient to improve function in preclinical muscular dystrophy. ESC Heart Fail. 2020, 7, 3983–3995. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Early right ventricular fibrosis and reduction in biventricular cardiac reserve in the dystrophin-deficient mdx heart. Am. J. Physiol. Circ. Physiol. 2015, 308, H303–H315. [Google Scholar] [CrossRef]
- Bauer, R.; Straub, V.; Blain, A.; Bushby, K.; MacGowan, G.A. Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 463–471. [Google Scholar] [CrossRef]
- Rafael-Fortney, J.A.; Chimanji, N.S.; Schill, K.E.; Martin, C.D.; Murray, J.D.; Ganguly, R.; Stangland, J.E.; Tran, T.; Xu, Y.; Canan, B.D.; et al. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 2011, 124, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Bish, L.T.; Yarchoan, M.; Sleeper, M.M.; Gazzara, J.A.; Morine, K.J.; Acosta, P.; Barton, E.R.; Sweeney, H.L. Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS ONE 2011, 6, e20856. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Cabrera, D.; Cespedes, C.; Vio, C.P.; Vazquez, Y.; Brandan, E.; Cabello-Verrugio, C. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. 2013, 353, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Verrugio, C.; Cordova, G.; Salas, J.D. Angiotensin II: Role in skeletal muscle atrophy. Curr. Protein Pept. Sci. 2012, 13, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Laurino, A.; Spinelli, V.; Gencarelli, M.; Balducci, V.; Dini, L.; Diolaiuti, L.; Ghionzoli, M.; Messineo, A.; Mugelli, A.; Cerbai, E.; et al. Angiotensin-II Drives Human Satellite Cells Toward Hypertrophy and Myofibroblast Trans-Differentiation by Two Independent Pathways. Int. J. Mol. Sci. 2019, 20, 4192. [Google Scholar] [CrossRef] [PubMed]
- Cozzoli, A.; Nico, B.; Sblendorio, V.T.; Capogrosso, R.F.; Dinardo, M.M.; Longo, V.; Gagliardi, S.; Montagnani, M.; De Luca, A. Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol. Res. 2011, 64, 482–492. [Google Scholar] [CrossRef]
- Peterson, J.M.; Wang, D.J.; Shettigar, V.; Roof, S.R.; Canan, B.D.; Bakkar, N.; Shintaku, J.; Gu, J.M.; Little, S.C.; Ratnam, N.M.; et al. NF-kappaB inhibition rescues cardiac function by remodeling calcium genes in a Duchenne muscular dystrophy model. Nat. Commun. 2018, 9, 3431. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. JASN 2010, 21, 93–102. [Google Scholar] [CrossRef]
- Cleutjens, J.P.; Verluyten, M.J.; Smiths, J.F.; Daemen, M.J. Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 1995, 147, 325–338. [Google Scholar]
- Whittaker, P.; Kloner, R.A.; Przyklenk, K. Intramyocardial injections and protection against myocardial ischemia. An attempt to examine the cardioprotective actions of adenosine. Circulation 1996, 93, 2043–2057. [Google Scholar] [CrossRef]
- Fomovsky, G.M.; Rouillard, A.D.; Holmes, J.W. Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J. Mol. Cell. Cardiol. 2012, 52, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, Q.; Wang, K.; Wang, S.; Lu, D.; Li, Z.; Geng, J.; Fang, P.; Wang, Y.; Shan, Q. Renal Denervation Findings on Cardiac and Renal Fibrosis in Rats with Isoproterenol Induced Cardiomyopathy. Sci. Rep. 2015, 5, 18582. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Li, C.; Han, R.; Kang, L.; Wang, J.; Gao, Y.; Li, Y.; He, J.; Tian, J. Pirfenidone controls the feedback loop of the AT1R/p38 MAPK/renin-angiotensin system axis by regulating liver X receptor-alpha in myocardial infarction-induced cardiac fibrosis. Sci. Rep. 2017, 7, 40523. [Google Scholar] [CrossRef]
- Sano, M.; Fukuda, K.; Sato, T.; Kawaguchi, H.; Suematsu, M.; Matsuda, S.; Koyasu, S.; Matsui, H.; Yamauchi-Takihara, K.; Harada, M.; et al. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 2001, 89, 661–669. [Google Scholar] [CrossRef]
- George, M.; Vijayakumar, A.; Dhanesh, S.B.; James, J.; Shivakumar, K. Molecular basis and functional significance of Angiotensin II-induced increase in Discoidin Domain Receptor 2 gene expression in cardiac fibroblasts. J. Mol. Cell. Cardiol. 2016, 90, 59–69. [Google Scholar] [CrossRef]
- Olson, E.R.; Shamhart, P.E.; Naugle, J.E.; Meszaros, J.G. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension 2008, 51, 704–711. [Google Scholar] [CrossRef]
- Su, X.; Shen, Y.; Jin, Y.; Weintraub, N.L.; Tang, Y. Identification of critical molecular pathways involved in exosome-mediated improvement of cardiac function in a mouse model of muscular dystrophy. Acta Pharmacol. Sin. 2020, 1–7. [Google Scholar] [CrossRef]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef]
- Allen, R.E.; Boxhorn, L.K. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J. Cell. Physiol. 1987, 133, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Foster, W.; Deasy, B.M.; Chan, Y.; Prisk, V.; Tang, Y.; Cummins, J.; Huard, J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: A key event in muscle fibrogenesis. Am. J. Pathol. 2004, 164, 1007–1019. [Google Scholar] [CrossRef]
- Ceco, E.; McNally, E.M. Modifying muscular dystrophy through transforming growth factor-beta. FEBS J. 2013, 280, 4198–4209. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, P.; Torchiana, E.; Confalonieri, P.; Brugnoni, R.; Barresi, R.; Mora, M.; Cornelio, F.; Morandi, L.; Mantegazza, R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Investig. 1995, 96, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Chiba, Y.; Uematsu, M.; Hino-Fukuyo, N.; Tanaka, S.; Onuma, A.; Iinuma, K.; Tsuchiya, S. Elevated plasma levels of tissue inhibitors of metalloproteinase-1 and their overexpression in muscle in human and mouse muscular dystrophy. J. Neurol. Sci. 2010, 297, 19–28. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 816–827. [Google Scholar]
- Au, C.G.; Butler, T.L.; Sherwood, M.C.; Egan, J.R.; North, K.N.; Winlaw, D.S. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int. J. Exp. Pathol. 2011, 92, 57–65. [Google Scholar] [CrossRef]
- Ieronimakis, N.; Hays, A.L.; Janebodin, K.; Mahoney, W.M., Jr.; Duffield, J.S.; Majesky, M.W.; Reyes, M. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFbeta1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 2013, 63, 122–134. [Google Scholar] [CrossRef]
- Spurney, C.F.; Knoblach, S.; Pistilli, E.E.; Nagaraju, K.; Martin, G.R.; Hoffman, E.P. Dystrophin-deficient cardiomyopathy in mouse: Expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul. Disord. NMD 2008, 18, 371–381. [Google Scholar] [CrossRef]
- Fogagnolo Mauricio, A.; Pereira, J.A.; Santo Neto, H.; Marques, M.J. Effects of fish oil containing eicosapentaenoic acid and docosahexaenoic acid on dystrophic mdx mice hearts at later stages of dystrophy. Nutrition 2016, 32, 855–862. [Google Scholar] [CrossRef]
- Ballmann, C.; Hollinger, K.; Selsby, J.T.; Amin, R.; Quindry, J.C. Histological and biochemical outcomes of cardiac pathology in mdx mice with dietary quercetin enrichment. Exp. Physiol. 2015, 100, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Barbin, I.C.; Pereira, J.A.; Bersan Rovere, M.; de Oliveira Moreira, D.; Marques, M.J.; Santo Neto, H. Diaphragm degeneration and cardiac structure in mdx mouse: Potential clinical implications for Duchenne muscular dystrophy. J. Anat. 2016, 228, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.A.; Bogdanovich, S.; Beiriger, A.; Wren, L.M.; Rossi, A.E.; Gao, Q.Q.; Gardner, B.B.; Earley, J.U.; Molkentin, J.D.; McNally, E.M. Excess SMAD signaling contributes to heart and muscle dysfunction in muscular dystrophy. Hum. Mol. Genet. 2014, 23, 6722–6731. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.A.; Kelly, S.M.; LoPresti, P.P.; Heydemann, A.; Earley, J.U.; Ferguson, E.L.; Wolf, M.J.; McNally, E.M. SMAD signaling drives heart and muscle dysfunction in a Drosophila model of muscular dystrophy. Hum. Mol. Genet. 2011, 20, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.D.; Jassal, D.S.; Halevy, O.; Pines, M.; Anderson, J.E. Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am. J. Physiol. Circ. Physiol. 2008, 294, H1550–H1561. [Google Scholar] [CrossRef]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef]
- Lee, E.M.; Kim, D.Y.; Kim, A.Y.; Lee, E.J.; Kim, S.H.; Lee, M.M.; Sung, S.E.; Park, J.K.; Jeong, K.S. Chronic effects of losartan on the muscles and the serologic profiles of mdx mice. Life Sci. 2015, 143, 35–42. [Google Scholar] [CrossRef]
- He, Z.; Way, K.J.; Arikawa, E.; Chou, E.; Opland, D.M.; Clermont, A.; Isshiki, K.; Ma, R.C.; Scott, J.A.; Schoen, F.J.; et al. Differential regulation of angiotensin II-induced expression of connective tissue growth factor by protein kinase C isoforms in the myocardium. J. Biol. Chem. 2005, 280, 15719–15726. [Google Scholar] [CrossRef]
- Gu, J.; Liu, X.; Wang, Q.X.; Tian, H.-W.; Guo, M.; Jiang, W.; Zhou, L. Angiotensin II increases CTGF expression via MAPKs/TGF-beta1/TRAF6 pathway in atrial fibroblasts. Exp. Cell Res. 2012, 318, 2105–2115. [Google Scholar] [CrossRef]
- Mondorf, U.F.; Geiger, H.; Herrero, M.; Zeuzem, S.; Piiper, A. Involvement of the platelet-derived growth factor receptor in angiotensin II-induced activation of extracellular regulated kinases 1 and 2 in human mesangial cells. FEBS Lett. 2000, 472, 129–132. [Google Scholar] [CrossRef]
- Soslow, J.H.; Xu, M.; Slaughter, J.C.; Crum, K.; Chew, J.D.; Burnette, W.B.; Su, Y.R.; Tomasek, K.; Parra, D.A.; Markham, L.W. The Role of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Duchenne Muscular Dystrophy Cardiomyopathy. J. Card. Fail. 2019, 25, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, M.; Hu, Z.; Hu, S.; Zou, Q.; Lai, X.; Liu, B.; Jiang, H.; Huang, C.; Wu, G. Angiotensin II Facilitates Matrix Metalloproteinase-9-Mediated Myosin Light Chain Kinase Degradation in Pressure Overload-Induced Cardiac Hypertrophy. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 2281–2295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, C.; Hong, S.; Min, J.; Yang, Q.; Hu, M.; Zhao, Y.; Hong, L. Excess mechanical stress and hydrogen peroxide remodel extracellular matrix of cultured human uterosacral ligament fibroblasts by disturbing the balance of MMPs/TIMPs via the regulation of TGFbeta1 signaling pathway. Mol. Med. Rep. 2017, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Schnee, J.; Wang, W.; Kim, S.; Fishbein, M.C.; Bruemmer, D.; Law, R.E.; Nicholas, S.; Ross, R.S.; Hsueh, W.A. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J. Am. Coll. Cardiol. 2004, 43, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Zhou, S.X.; Lei, J.; Yuan, G.Y.; Wang, J.F. Blockades of angiotensin and aldosterone reduce osteopontin expression and interstitial fibrosis infiltration in rats with myocardial infarction. Chin. Med. J. 2008, 121, 2192–2196. [Google Scholar] [CrossRef]
- Dahiya, S.; Givvimani, S.; Bhatnagar, S.; Qipshidze, N.; Tyagi, S.C.; Kumar, A. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol. 2011, 187, 2723–2731. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, T.; Chen, Y.; Ahokas, R.A.; Sun, Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol. Cell. Biochem. 2008, 317, 43–50. [Google Scholar] [CrossRef]
- Takenaka, H.; Kihara, Y.; Iwanaga, Y.; Onozawa, Y.; Toyokuni, S.; Kita, T. Angiotensin II, oxidative stress, and extracellular matrix degradation during transition to LV failure in rats with hypertension. J. Mol. Cell. Cardiol. 2006, 41, 989–997. [Google Scholar] [CrossRef]
- Kakishita, M.; Nakamura, K.; Asanuma, M.; Morita, H.; Saito, H.; Kusano, K.; Nakamura, Y.; Emori, T.; Matsubara, H.; Sugaya, T.; et al. Direct evidence for increased hydroxyl radicals in angiotensin II-induced cardiac hypertrophy through angiotensin II type 1a receptor. J. Cardiovasc. Pharmacol. 2003, 42 (Suppl. 1), S67–S70. [Google Scholar] [CrossRef]
- Lu, L.; Quinn, M.T.; Sun, Y. Oxidative stress in the infarcted heart: Role of de novo angiotensin II production. Biochem. Biophys. Res. Commun. 2004, 325, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Griendling, K.K. NADPH oxidases and angiotensin II receptor signaling. Mol. Cell. Endocrinol. 2009, 302, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Somanna, N.K.; Valente, A.J.; Krenz, M.; Fay, W.P.; Delafontaine, P.; Chandrasekar, B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. J. Cell. Physiol. 2016, 231, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Minas, J.N.; Thorwald, M.A.; Conte, D.; Vazquez-Medina, J.P.; Nishiyama, A.; Ortiz, R.M. Angiotensin and mineralocorticoid receptor antagonism attenuates cardiac oxidative stress in angiotensin II-infused rats. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1178–1188. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Colston, J.T.; de la Rosa, S.D.; Strader, J.R.; Anderson, M.A.; Freeman, G.L. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. Febs Lett. 2005, 579, 2533–2540. [Google Scholar] [CrossRef]
- Delfin, D.A.; Xu, Y.; Peterson, J.M.; Guttridge, D.C.; Rafael-Fortney, J.A.; Janssen, P.M. Improvement of cardiac contractile function by peptide-based inhibition of NF-kappaB in the utrophin/dystrophin-deficient murine model of muscular dystrophy. J. Transl. Med. 2011, 9, 68. [Google Scholar] [CrossRef]
- Ballmann, C.; Denney, T.; Beyers, R.J.; Quindry, T.; Romero, M.; Selsb, J.T.; Quindry, J.C. Long-term dietary quercetin enrichment as a cardioprotective countermeasure in mdx mice. Exp. Physiol. 2017, 102, 635–649. [Google Scholar] [CrossRef]
- Peng, J.; Gurantz, D.; Tran, V.; Cowling, R.T.; Greenberg, B.H. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ. Res. 2002, 91, 1119–1126. [Google Scholar] [CrossRef]
- Ermolova, N.V.; Martinez, L.; Vetrone, S.A.; Jordan, M.C.; Roos, K.P.; Sweeney, H.L.; Spencer, M.J. Long-term administration of the TNF blocking drug Remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul. Disord. NMD 2014, 24, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Mareedu, S.; Pachon, R.E.; Jayapalraj, T.; Fefelova, N.; Balakrishnan, R.; Niranjan, N.; Xie, L.H.; Babu, G.J. Sarcolipin haploinsufficiency prevents dystrophic cardiomyopathy in mdx mice. Am. J. Physiol. Circ. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sciorati, C.; Staszewsky, L.; Zambelli, V.; Russo, I.; Salio, M.; Novelli, D.; Di Grigoli, G.; Moresco, R.M.; Clementi, E.; Latini, R. Ibuprofen plus isosorbide dinitrate treatment in the mdx mice ameliorates dystrophic heart structure. Pharmacol. Res. 2013, 73, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Sriramula, S.; Francis, J. Tumor Necrosis Factor-Alpha Is Essential for Angiotensin II-Induced Ventricular Remodeling: Role for Oxidative Stress. PLoS ONE 2015, 10, e0138372. [Google Scholar] [CrossRef]
- Farini, A.; Gowran, A.; Bella, P.; Sitzia, C.; Scopece, A.; Castiglioni, E.; Rovina, D.; Nigro, P.; Villa, C.; Fortunato, F.; et al. Fibrosis Rescue Improves Cardiac Function in Dystrophin-Deficient Mice and Duchenne Patient-Specific Cardiomyocytes by Immunoproteasome Modulation. Am. J. Pathol. 2019, 189, 339–353. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.X.; Han, Q.Y.; Li, W.J.; Zhang, Y.L.; Du, J.; Xia, Y.L.; Li, H.H. Activation of the cardiac proteasome promotes angiotension II-induced hypertrophy by down-regulation of ATRAP. J. Mol. Cell. Cardiol. 2015, 79, 303–314. [Google Scholar] [CrossRef]
- Blain, A.; Greally, E.; Laval, S.H.; Blamire, A.M.; MacGowan, G.A.; Straub, V.W. Absence of Cardiac Benefit with Early Combination ACE Inhibitor and Beta Blocker Treatment in mdx Mice. J. Cardiovasc. Transl. Res. 2015, 8, 198–207. [Google Scholar] [CrossRef]
- Lee, E.M.; Kim, A.Y.; Lee, E.J.; Park, J.K.; Lee, M.M.; Hwang, M.; Kim, C.Y.; Kim, S.Y.; Jeong, K.S. Therapeutic effects of mouse adipose-derived stem cells and losartan in the skeletal muscle of injured mdx mice. Cell Transplant. 2015, 24, 939–953. [Google Scholar] [CrossRef]
- Meyers, T.A.; Heitzman, J.A.; Krebsbach, A.M.; Aufdembrink, L.M.; Hughes, R.; Bartolomucci, A.; Townsend, D. Acute AT1R blockade prevents isoproterenol-induced injury in mdx hearts. J. Mol. Cell. Cardiol. 2019, 128, 51–61. [Google Scholar] [CrossRef]
- Heier, C.R.; Yu, Q.; Fiorillo, A.A.; Tully, C.B.; Tucker, A.; Mazala, D.A.; Uaesoontrachoon, K.; Srinivassane, S.; Damsker, J.M.; Hoffman, E.P.; et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci. Alliance 2019, 2, e201800186. [Google Scholar] [CrossRef]
- Lowe, J.; Floyd, K.T.; Rastogi, N.; Schultz, E.J.; Chadwick, J.A.; Swager, S.A.; Zins, J.G.; Kadakia, F.K.; Smart, S.; Gomez-Sanchez, E.P.; et al. Similar efficacy from specific and non-specific mineralocorticoid receptor antagonist treatment of muscular dystrophy mice. J. Neuromuscul. Dis. 2016, 3, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Kadakia, F.K.; Zins, J.G.; Haupt, M.; Peczkowski, K.K.; Rastogi, N.; Floyd, K.T.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E.; Elnakish, M.T.; et al. Mineralocorticoid Receptor Antagonists in Muscular Dystrophy Mice During Aging and Exercise. J. Neuromuscul. Dis. 2018, 5, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.M.; Murray, J.D.; Schill, K.E.; Rastogi, N.; Schultz, E.J.; Tran, T.; Raman, S.V.; Rafael-Fortney, J.A. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLoS ONE 2014, 9, e88360. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Verrugio, C.; Morales, M.G.; Cabrera, D.; Vio, C.P.; Brandan, E. Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J. Cell. Mol. Med. 2012, 16, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 2195–2204. [Google Scholar] [CrossRef]
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220. [Google Scholar] [CrossRef]
- Bostick, B.; Yue, Y.; Long, C.; Duan, D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ. Res. 2008, 102, 121–130. [Google Scholar] [CrossRef]
- Bostick, B.; Yue, Y.; Long, C.; Marschalk, N.; Fine, D.M.; Chen, J.; Duan, D. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged Mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 253–261. [Google Scholar] [CrossRef]
- Hakim, C.H.; Grange, R.W.; Duan, D. The passive mechanical properties of the extensor digitorum longus muscle are compromised in 2- to 20-mo-old mdx mice. J. Appl. Physiol. 2011, 110, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.S.; Hinkle, R.T.; Chamberlain, J.S.; Brooks, S.V.; Faulkner, J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6-28 months old. J. Physiol. 2001, 535, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- van Putten, M.; Kumar, D.; Hulsker, M.; Hoogaars, W.M.; Plomp, J.J.; van Opstal, A.; van Iterson, M.; Admiraal, P.; van Ommen, G.J.; AC’t Hoen, P.A.; et al. Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains. Neuromuscul. Disord. Nmd 2012, 22, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Bao, B.; Echigoya, Y.; Yokota, T. Dystrophin-deficient large animal models: Translational research and exon skipping. Am. J. Transl. Res. 2015, 7, 1314–1331. [Google Scholar] [PubMed]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 2018, 3, 4. [Google Scholar] [CrossRef]
- Rovina, D.; Castiglioni, E.; Niro, F.; Mallia, S.; Pompilio, G.; Gowran, A. “Betwixt Mine Eye and Heart a League Is Took”: The Progress of Induced Pluripotent Stem-Cell-Based Models of Dystrophin-Associated Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 6997. [Google Scholar] [CrossRef]
- Jelinkova, S.; Vilotic, A.; Pribyl, J.; Aimond, F.; Salykin, A.; Acimovic, I.; Pesl, M.; Caluori, G.; Klimovic, S.; Urban, T.; et al. DMD Pluripotent Stem Cell Derived Cardiac Cells Recapitulate in vitro Human Cardiac Pathophysiology. Front. Bioeng. Biotechnol. 2020, 8, 535. [Google Scholar] [CrossRef]
- Pioner, J.M.; Fornaro, A.; Coppini, R.; Ceschia, N.; Sacconi, L.; Donati, M.A.; Favilli, S.; Poggesi, C.; Olivotto, I.; Ferrantini, C. Advances in Stem Cell Modeling of Dystrophin-Associated Disease: Implications for the Wider World of Dilated Cardiomyopathy. Front. Physiol. 2020, 11, 368. [Google Scholar] [CrossRef]
- Bourke, J.P.; Bueser, T.; Quinlivan, R. Interventions for preventing and treating cardiac complications in Duchenne and Becker muscular dystrophy and X-linked dilated cardiomyopathy. Cochrane Database Syst. Rev. 2018, 10, CD009068. [Google Scholar] [CrossRef]
- Hor, K.N.; Mazur, W.; Taylor, M.D.; Al-Khalidi, H.R.; Cripe, L.H.; Jefferies, J.L.; Raman, S.V.; Chung, E.S.; Kinnett, K.J.; Williams, K.; et al. Effects of steroids and angiotensin converting enzyme inhibition on circumferential strain in boys with Duchenne muscular dystrophy: A cross-sectional and longitudinal study utilizing cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson. 2011, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Cardona, A.; He, X.; Halnon, N.; Markham, L.; Soslow, J.H.; Puchalski, M.D.; Auerbach, S.R.; et al. Stabilization of Early Duchenne Cardiomyopathy with Aldosterone Inhibition: Results of the Multicenter AIDMD Trial. J. Am. Hear. Assoc. 2019, 8, e013501. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Halnon, N.J.; Kissel, J.T.; He, X.; Tran, T.; Smart, S.; McCarthy, B.; Taylor, M.D.; et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 153–161. [Google Scholar] [CrossRef]
- Raman, S.V.; Hor, K.N.; Mazur, W.; He, X.; Kissel, J.T.; Smart, S.; McCarthy, B.; Roble, S.L.; Cripe, L.H. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: Results of a two-year open-label extension trial. Orphanet J. Rare Dis. 2017, 12, 39. [Google Scholar] [CrossRef]
- Duboc, D.; Meune, C.; Pierre, B.; Wahbi, K.; Eymard, B.; Toutain, A.; Berard, C.; Vaksmann, G.; Weber, S.; Becane, H.M. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am. Heart J. 2007, 154, 596–602. [Google Scholar] [CrossRef]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef]
- Duboc, D.; Meune, C.; Lerebours, G.; Devaux, J.Y.; Vaksmann, G.; Becane, H.M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2005, 45, 855–857. [Google Scholar] [CrossRef]
- Ramaciotti, C.; Heistein, L.C.; Coursey, M.; Lemler, M.S.; Eapen, R.S.; Iannaccone, S.T.; Scott, W.A. Left ventricular function and response to enalapril in patients with duchenne muscular dystrophy during the second decade of life. Am. J. Cardiol. 2006, 98, 825–827. [Google Scholar] [CrossRef]
- Kajimoto, H.; Ishigaki, K.; Okumura, K.; Tomimatsu, H.; Nakazawa, M.; Saito, K.; Osawa, M.; Nakanishi, T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ. J. Off. J. Jpn. Circ. Soc. 2006, 70, 991–994. [Google Scholar] [CrossRef]
- Ogata, H.; Ishikawa, Y.; Ishikawa, Y.; Minami, R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J. Cardiol. 2009, 53, 72–78. [Google Scholar] [CrossRef]
- Kwon, H.W.; Kwon, B.S.; Kim, G.B.; Chae, J.H.; Park, J.D.; Bae, E.J.; Noh, C.I. The effect of enalapril and carvedilol on left ventricular dysfunction in middle childhood and adolescent patients with muscular dystrophy. Korean Circ. J. 2012, 42, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.D.; Flanigan, K.M.; Thrush, P.T.; Dvorchik, I.; Yin, H.; Canter, C.; Connolly, A.M.; Parrish, M.; McDonald, C.M.; Braunlin, E.; et al. A randomized, double-blind trial of lisinopril and losartan for the treatment of cardiomyopathy in duchenne muscular dystrophy. PLoS Curr. 2013, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Magalhaes, T.A.; Meira, Z.M.; Rassi, C.H.; Andrade, A.C.; Gutierrez, P.S.; Azevedo, C.F.; Gurgel-Giannetti, J.; Vainzof, M.; Zatz, M.; et al. Myocardial Fibrosis Progression in Duchenne and Becker Muscular Dystrophy: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 190–199. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | DMD | BMD |
|---|---|---|
| Genetic defect | Out-of-frame mutation inXp21.1 chromosome | In-frame mutation in Xp21.1 chromosome |
| Dystrophin protein | Absent | Present but partially functional |
| Prevalence | 1/3500–5000 male births | 1/18,000–20,000 male births |
| Age at diagnosis | 3–6 years | 10–14 years |
| Non-ambulatory phase | 12–14 years | 30s |
| Life expectancy | 20–30 s | 40–50 s |
| Prevalence of DDC | Approximately 100% | 50% |
| Clinically evident DDC | 15–18 years; always after skeletal muscle symptoms | Variable; not related with skeletal muscle symptoms |
| Histological hallmark | Cardiac Fibrosis | |
| Leading cause of death | Cardiac (end-stage CHF or VA) | |
| Genotype | Life Expectancy | DDC Age of Onset | Histopathology | Echocardiographic Changes |
|---|---|---|---|---|
| Wild tipe | 2 years | None | Normal | None |
| mdx | 1.5–2 years | 10 months | Mild | Mild/none |
| mdx/Utr | 20 weeks | 8 weeks | Moderate | Moderate |
| mdx/Dtna | 8–10 months | 4 weeks | Moderate/severe | - |
| mdx/7 | <4 weeks | 3 weeks (20 days) | Mild | None |
| mdx/Myod1 | 12 months | 5 months | Severe | - |
| mdx/Cmah | 11 months | 3 months | Moderate/severe | - |
| mdx/mTR G2 | 4–12 months | 32 weeks | Severe | Severe |
| Author | Type of Study | Size | Interventions | Outcomes |
|---|---|---|---|---|
| Hor et al. (2011) [192] | Retrospective cohort study | DMD: 136 | - Deflazacort or prednisone and lisinopril or enalapril or losartan: 92; Glucocorticoid alone: 114 | ACE-I/ARB therapy combined with glucocorticoids did not arrest the decline in cardiac function. |
| Raman et al. (2019) [193] | Double-blind, randomized, noninferiority trial | DMD: 52 | - Eplerenone: 26/52; Spironolactone: 26/52 | Spironolactone added to background therapy is noninferior to eplerenone in preserving heart function. |
| Raman et al. (2015) [194] | Randomized, double-blind, placebo-controlled trial | DMD: 42 | - Eplerenone: 20/42; Placebo: 22/42 | Eplerenone added to ACEI or ARB therapy attenuates the progressive decline ventricular function. |
| Raman et al. (2017) [195] | Randomized, double-blind, placebo-controlled trial | DMD: 11 | - Eplerenone: Placebo | Eplerenone is a useful if is initiated in the first phases with no relevant dysfunction. |
| Duboc et al. (2007) [196] | Randomized Control Trial | DMD: 57 | - Phase I (3 years): 56/57 Perindopril or Placebo. - Phase II (2 years): 51/57 Perindopril | Phase I: improvement of ventricular function in 55/56 patients. Phase I and II: Early treatment with perindopril delayed the onset and progression of prominent left ventricle dysfunction. |
| Jefferies et al. (2005) [197] | Retrospective case series. | DMD: 62; BMD: 7 | -ACE inhibitors: 13/31; ACE inhibitor and β-blocker: 18/31 | 2/29: showed no deterioration in LV function. 8/29: showed improvement in LV size or function or both. 19/29: showed normalization of LV size or function or both. |
| Duboc et al. (2005) [198] | Randomized Control Trial | DMD: 57 | - Placebo: 29/57; Perindopril: 28/57 | Early initiation of treatment with perindopril is associated with a lower mortality in patients with DMD with normal LV ejection fraction at study entry. |
| Ramaciotti et al. (2006) [199] | Retrospective case series | DMD: 50 | - Enalapril. | 10/26 (43%) presented improvement with the use of enalapril normalizing the shortening fraction. |
| Kajimoto et al. (2006) [200] | Randomized Control Trial | DMD: 25; FMD: 2 EDMD: 1 | - Enalapril or Cilazapril and Carvedilol: 13/28 - ACE-I alone: 15/28 | No significant change was observed in patients who received ACE-I monotherapy. Carvedilol plus an ACEI improves left ventricular systolic function in patients with muscular dystrophy. |
| Ogata et al. (2009) [201] | Retrospective cohort study | DMD: 52 | - Enalapril or Lisinopril and Bisoprolol or Metoprolol or Cavedilol | In DMD patients with heart failure the combination of an ACE inhibitor and a beta-blocker had a beneficial effect on survival. |
| Kwon et al. (2012) [202] | Retrospective cohort study | DMD: 22; BMD: 1 | - Enalapril 13/23; Carvedilol: 10/23 | Carvedilol or Enalapril could improve LV systolic function in patients with muscular dystrophy. |
| Viollet et al. (2012) [203] | Retrospective cohort study | DMD: 42 | - Lisinopril and metoprolol/atenolol; Lisinopril. | Treatment with ACE inhibitor or ACE inhibitor plus BB can delay progression of cardiomyopathy. |
| Allen et al. (2013) [204] | Randomized Control Trial | DMD: 23 | - Losartan: 11/23; Lisinopril: 12/23; | LV Ejection fraction improved equally with two difference therapeutic. |
| Silva et al. (2016) [205] | Randomized Control Trial | DMD: 70; BMD: 6 | - Placebo: 21/76; Enalapril: 21/76 | ACEI slows Myocardial fibrosis progression at a 2-year follow-up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Gonzalez, M.; Lubian-Gutierrez, M.; Cascales-Poyatos, H.M.; Perez-Reviriego, A.A.; Castellano-Martinez, A. Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 356. https://doi.org/10.3390/ijms22010356
Rodriguez-Gonzalez M, Lubian-Gutierrez M, Cascales-Poyatos HM, Perez-Reviriego AA, Castellano-Martinez A. Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy. International Journal of Molecular Sciences. 2021; 22(1):356. https://doi.org/10.3390/ijms22010356
Chicago/Turabian StyleRodriguez-Gonzalez, Moises, Manuel Lubian-Gutierrez, Helena Maria Cascales-Poyatos, Alvaro Antonio Perez-Reviriego, and Ana Castellano-Martinez. 2021. "Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy" International Journal of Molecular Sciences 22, no. 1: 356. https://doi.org/10.3390/ijms22010356
APA StyleRodriguez-Gonzalez, M., Lubian-Gutierrez, M., Cascales-Poyatos, H. M., Perez-Reviriego, A. A., & Castellano-Martinez, A. (2021). Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy. International Journal of Molecular Sciences, 22(1), 356. https://doi.org/10.3390/ijms22010356