1. Introduction
Hearing loss is the most prevalent form of sensory impairment, affecting about 466 million people worldwide including 34 million children [
1]. Most of the hearing loss is sensorineural due to disease, degeneration, or trauma to the cochlea of the inner ear [
2]. Treatment options for sensorineural hearing loss (SNHL) are currently limited to prosthetic devices such as hearing aids and cochlear implants. Both devices can partly restore auditory function, but these have limitations because the ear remains damaged. There is thus a significant need for the development of effective therapies to prevent cochlear damage and hearing loss or restore cochlear sensorineural structure and hearing. We have identified that signalling pathways activated by adenosine receptors are important regulators of cellular responses to injury in cochlear tissues. Animal studies reveal that stimulation of the A
1 adenosine receptor (A
1R) is particularly promising for the treatment of acute noise-induced cochlear injury [
3,
4] and other forms of SNHL such as from cytotoxic drugs, including cisplatin and aminoglycoside antibiotics [
5,
6]. The principal advantage of this approach is that the A
1R stimulation affects multiple mechanisms of cochlear injury (e.g., oxidative stress, glutamate excitotoxicity, activation of apoptotic pathways), thus providing comprehensive protection from SNHL [
7]. One of the issues with advancing these approaches to clinical applicability is the delivery of receptor agonists to the inner ear and their suitability for long-term therapy.
As an alternative to the use of exogenous A
1R agonists, we have previously considered other strategies that would regulate the action of endogenous adenosine, for example by manipulating intracellular adenosine metabolism [
8]. Recently, we have identified a novel otoprotective paradigm based on increasing A
1R responsiveness to endogenous adenosine, which can be achieved by inhibiting the Regulator of G protein Signalling 4 (RGS4). RGS is a large family of proteins that inhibit signal transduction pathways initiated by G protein-coupled receptors (GPCR) including A
1R [
9,
10,
11]. RGS increase the intrinsic GTPase activity of G proteins and thus enhance G protein inactivation and promote receptor desensitisation [
9].
In the past two decades, RGS proteins have received increasing interest as potential drug targets in cardiovascular disease, CNS disorders and several types of cancer [
12,
13,
14,
15]. Targeted inhibition of RGS proteins could potentially provide a way to fine-tune GPCR signalling by potentiating or prolonging the effect of receptor agonists. This approach could also be used to enhance endogenous ligand effects on GPCR. A few small molecule RGS inhibitors have been identified, particularly in the well-studied RGS4 family [
16,
17]. The selectivity of RGS proteins is mediated either through direct interaction with target receptors, or through selective interactions with accessory proteins [
16]. For example, the neurabin-RGS4 molecular complex regulates A
1R signalling events in the brain. Neurabin is an intracellular scaffolding protein (protein phosphatase 1 regulatory inhibitor subunit 9a, PPP1R9A) expressed in neural tissues which facilitates interactions of RGS4 with the A
1R [
18]. After A
1R stimulation by endogenous or exogenous ligands, neurabin forms a complex with RGS4 and recruits it to the cell surface to the A
1R [
18]. Disruption of the neurabin-RGS4 complex, either by genetic deletion of neurabin or by selective inhibitors of RGS4, enhances A
1R signalling even without administration of exogenous A
1R ligands. Mice with genetic deletion of neurabin are protected against kainate-induced seizures, evidenced by reduced severity and occurrence of seizures, improved neuronal survival and overall lower mortality [
18]. Similarly, this anticonvulsant and neuroprotective effect is also conferred by CCG-4986, a small molecule inhibitor of RGS4 [
18]. The neurabin/RGS4 complex thus appears to be the key regulator of A
1R activity in the brain and a promising neuroprotective target.
Here, we investigated an otoprotective strategy based on inhibition of the neurabin/RGS4 complex in the cochlea, with the aim to increase A1R responsiveness to endogenous adenosine released from cochlear tissues during acoustic stress. This novel otoprotective strategy is based on intratympanic injection of a small molecule RGS4 inhibitor to the round window membrane (RWM) of the cochlea.
3. Discussion
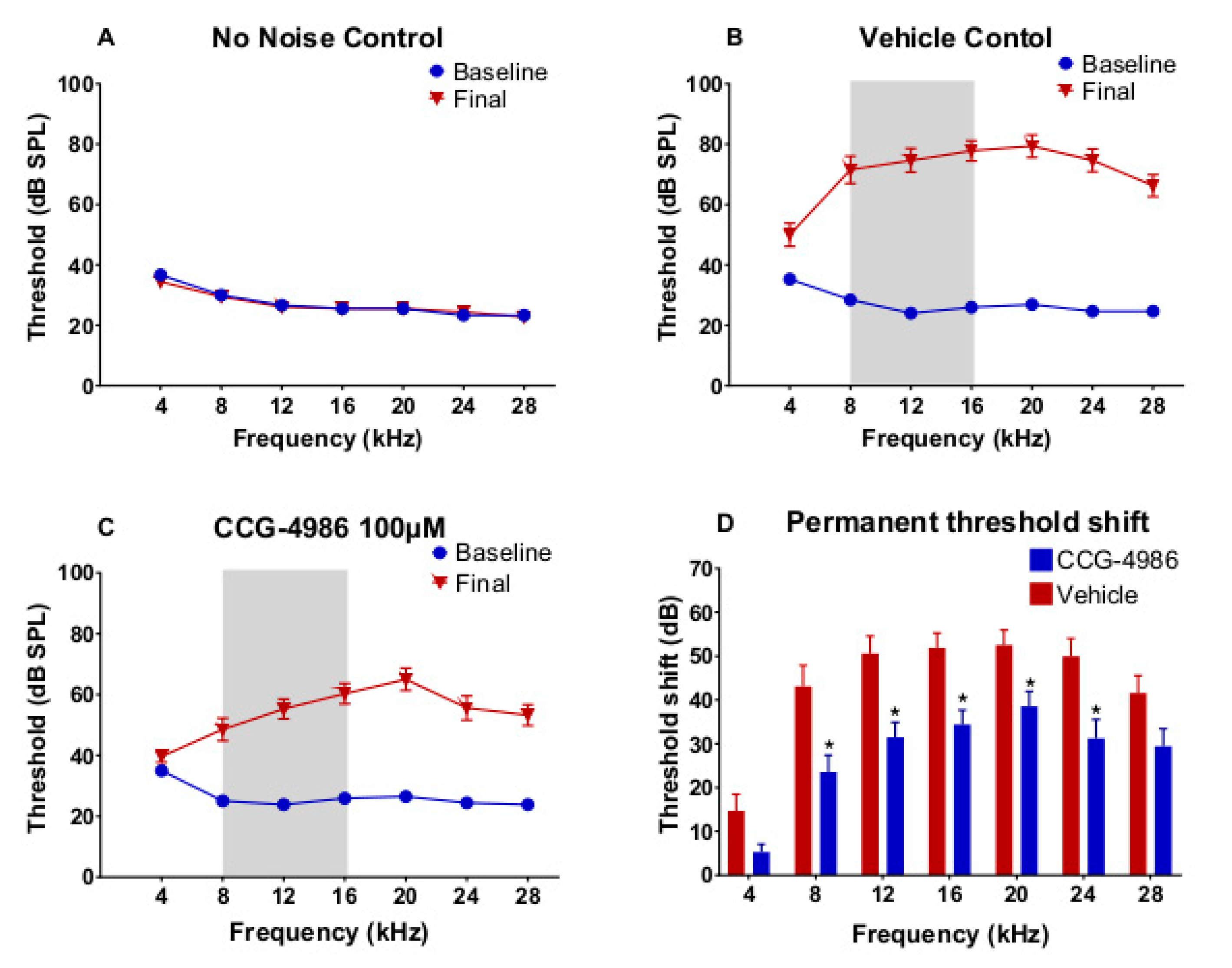
Our study demonstrates the cochlear rescue effect of CCG-4986 treatment in rats up to 48 h after traumatic noise exposure. This novel treatment is based on enhanced endogenous adenosine A
1R activity in the cochlea. CCG-4986 is a small molecule RGS4 inhibitor which disrupts the signalling complex (RGS4/Neurabin) that regulates A
1R activation [
18]. We have shown that immunolocalisation of this molecular complex in sensory hair cells and spiral ganglion neurons corresponds to A
1R distribution in the rat cochlea [
19,
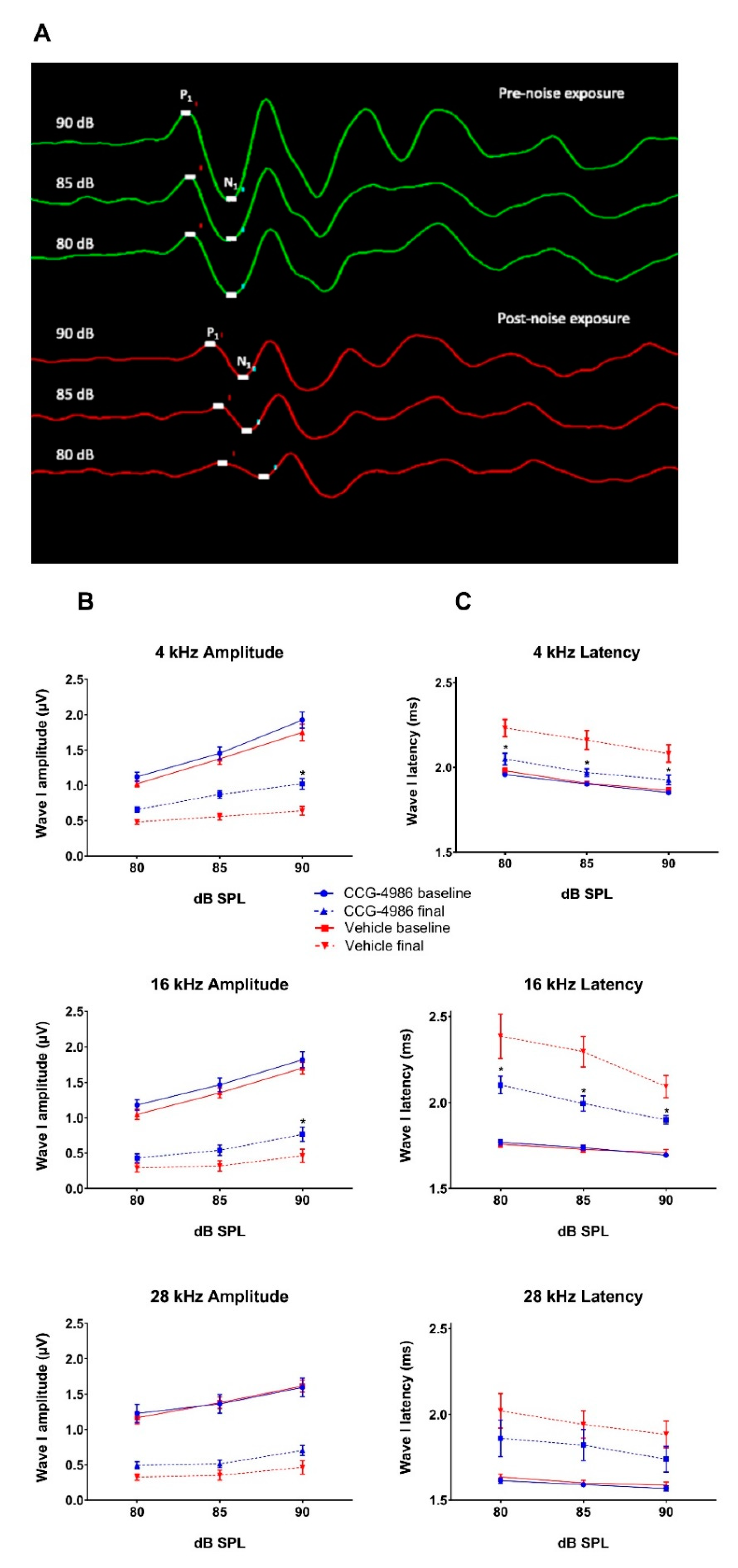
20]. Local intratympanic administration of CCG-4986 48 h after acoustic overexposure mitigated noise-induced threshold shifts by 10–19 dB, which is considered clinically significant. This otoprotective effect was greater when the CCG-4986 treatment was delivered earlier (24 h after noise exposure), effectively reducing moderate-severe hearing loss to mild hearing loss. CCG-4986 administration also improved suprathreshold responses in noise-exposed animals (increased amplitudes and reduced latencies of ABR wave I), suggesting partial recovery of auditory nerve function. The treatment enhanced the survival of sensory hair cells in the noise-exposed cochlea and mitigated noise-induced loss of afferent synapses in the frequency-specific regions. Noise-induced loss of spiral ganglion neurons was, however, irreversible. The present study thus demonstrates that the RGS4 inhibition is the promising strategy for the treatment of noise-induced cochlear injury and introduces a novel paradigm for the treatment of NIHL and other forms of SNHL based on regulation of GPCR.
3.1. Drug Delivery to the Inner Ear
The intratympanic method of drug delivery to the round window membrane has two principle advantages. Firstly, it precludes off-target effects of A
1R activation in cardiovascular and other tissues, which is an important caveat for systemic administration. Secondly, poloxamer-407 is liquid at low temperatures, but becomes a gel at body temperature which allows slow drug release to the cochlear perilymph [
21]. The small size of CCG-4986 (375 g/mol) and its apparent otoprotective effect suggest that this drug readily crosses into cochlear perilymph through the round window membrane. However, further studies are required to establish CCG-4986 concentration in cochlear fluid compartments after intratympanic injection and its pharmacokinetic properties.
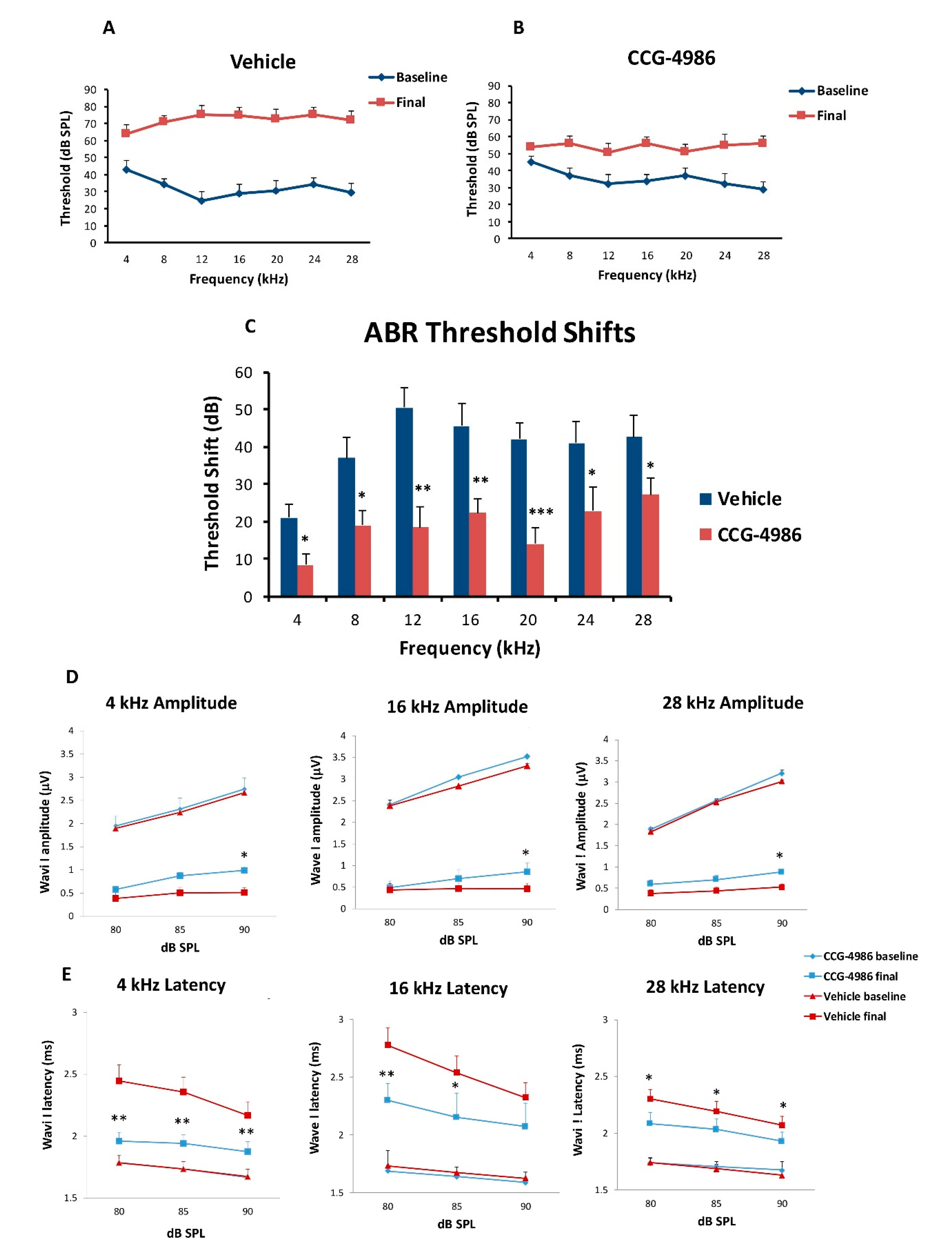
3.2. ABR Threshold Shifts and Suprathreshold Responses are Mitigated by CCG-4986
CCG-4986 treatment 48 h post-exposure produced a cochlear rescue effect by reducing PTS at all test frequencies above 4 kHz. The PTS reduction of >10 dB is considered clinically significant [
22]. As expected, the earlier treatment 24 h post-exposure enhanced the rescue effect of CCG-4986 (up to 32 dB at 12 kHz). The window of opportunity to treat NIHL was thus reminiscent of our previous study using an A
1R agonist adenosine amine congener [
23].
Whilst auditory thresholds are considered a good metric of hair cell function, they are poor indicator of neuronal damage in the cochlea [
24]. In this study, the changes in wave I amplitude and latency were used as indicators of afferent neural fibre (ANF) integrity. Noise overstimulation leads to a decrease in suprathreshold Wave 1 amplitudes [
25] and prolonged latencies [
26] due to reductions in synchronous firing, lower discharge rates, and decreased recruitment of the high threshold ANF [
27]. Recent studies postulate that the synaptopathy and the loss of ANF is a primary and mostly irreversible event in noise-induced hearing loss [
28]. This loss of cochlear afferents is functionally measured by reduced suprathreshold responses, due to preferential vulnerability of high threshold, low spontaneous rate ANF [
29]. The diffuse afferent denervation that cannot be detected by measuring auditory thresholds is thought to contribute to poor performance in complex auditory tasks such as speech discrimination in a noisy environment, and thus has been termed “hidden hearing loss” [
30]. In this study, treatment with CCG-4986 24 and 48 h after noise exposure reduced the ABR wave I latencies at all test frequencies, but only a minor improvement of wave I amplitudes was observed, suggesting partial recovery of neural injury.
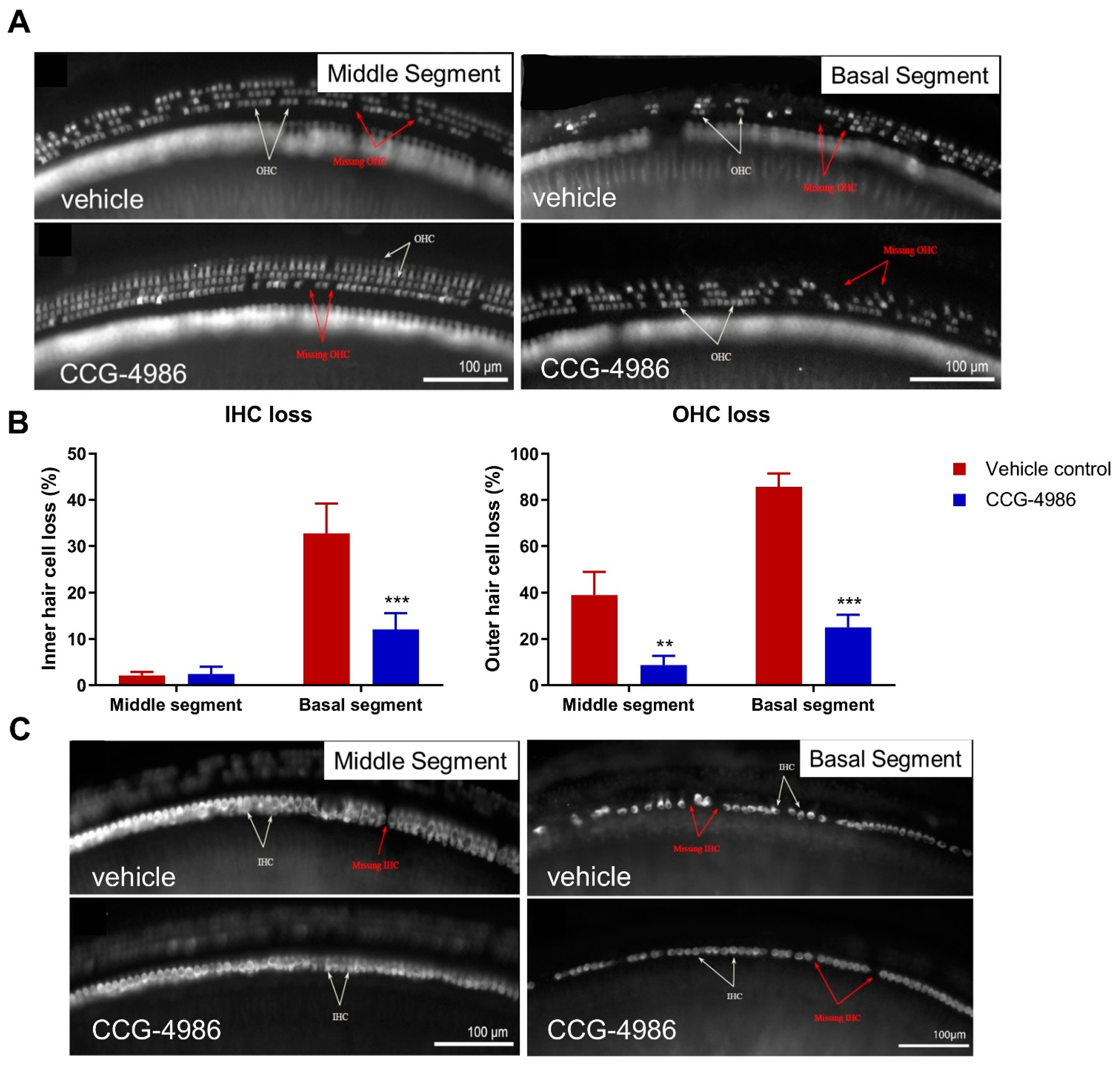
3.3. CCG-4986 Improves the Survival of Sensory Hair Cells
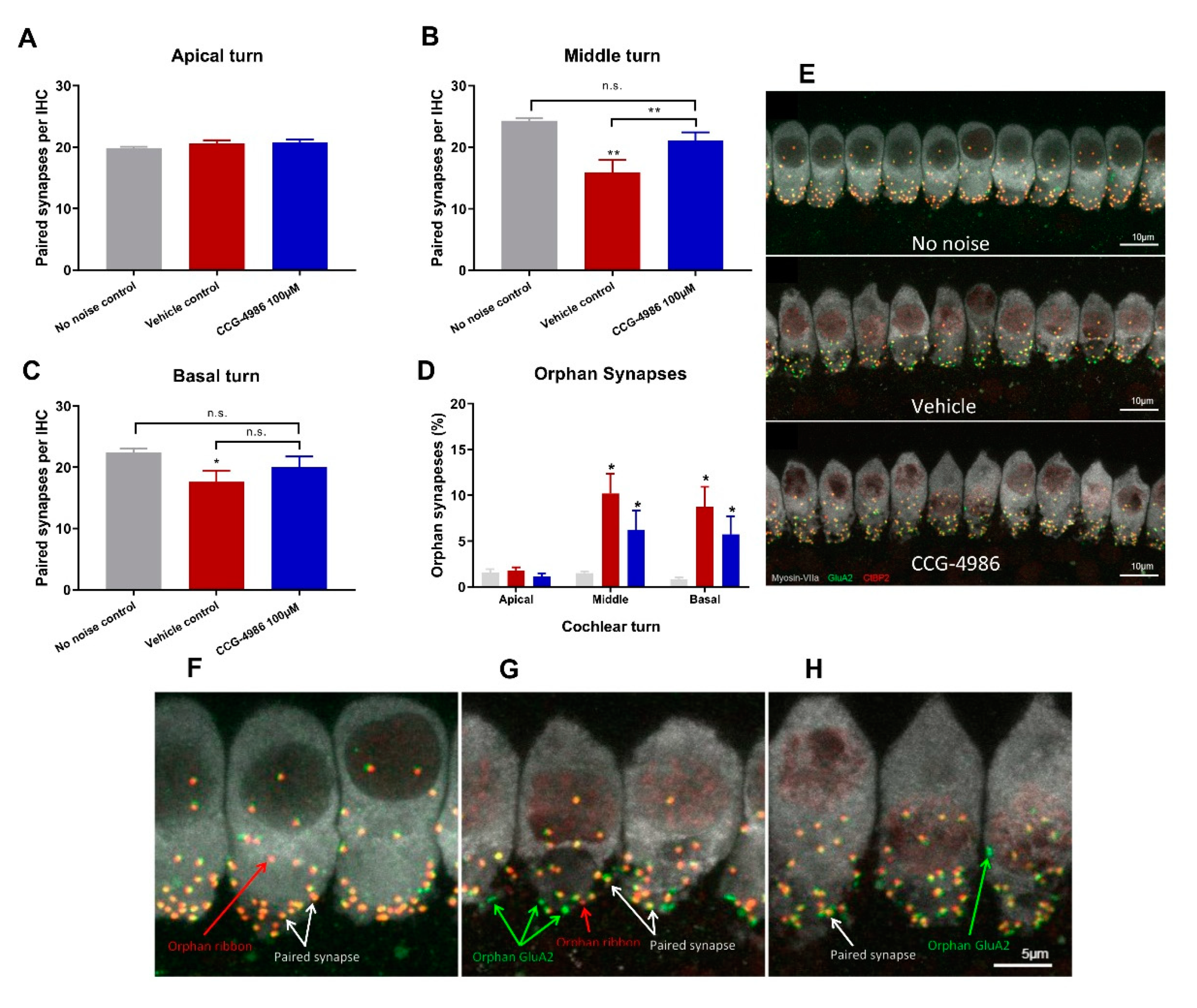
Auditory threshold shifts are largely determined by the integrity of sensory hair cells; hence, the quantitative histological analysis of hair cell survival was carried out in the apical, middle, and basal turns of the noise-exposed cochleae. The hair cell population in the apical segment was virtually unaffected by noise exposure, but a significant loss of OHC was observed in the middle and basal segments of the cochlea, the latter being most severely affected. There was very little IHC loss in the apical and middle turns, but almost one third of IHC was missing in the basal turn of the cochlea.
The increased vulnerability of OHC to noise, particularly in the basal turn, have been well documented in the past. "Inappropriate" loss of high frequency hair cells has also been observed in previous studies where the noise exposures targeted lower frequencies [
31]. Basal OHC are particularly vulnerable to acoustic insult, likely due to their lower antioxidant buffering capacity leading to increased susceptibility to oxidative stress [
32].
CCG-4986 treatment conferred a significant protection from noise-induced OHC loss in the middle segment and both IHC and OHC loss in the basal segment of the cochlea. Hair cell death is primarily mediated by oxidative stress and calcium overload leading to caspase-dependent cell death pathways [
33]. We postulate that the RGS4 inhibition by CCG-4986 enhanced endogenous adenosine A
1R signalling, which in turn improved antioxidant defences and restored calcium homeostasis [
3,
18].
3.4. CCG-4986 Partly Restores Afferent Synapses but does not Prevent Neuronal Loss
In the absence of noise exposure, the vast majority of IHC synapses contain a pre-synaptic ribbon paired with a post-synaptic terminal from a single ANF, and only around 1% of synapses appear as orphaned pre-synaptic ribbons or post-synaptic terminals [
25]. The survival of IHC-auditory nerve synapses is considered a sensitive metric for quantitative analysis of afferent innervation in the cochlea [
25].
Noise exposure affected only synaptic ribbons in the mid and high frequency regions, whilst IHC synapses in the low frequency region were mostly intact. Treatment with CCG-4986 yielded a robust neuroprotective effect in the middle turn, to such extent that the average number of functional (paired) synapses in drug-treated animals was not significantly different from control animals exposed to ambient noise.
IHC synaptic loss is an acute event that is usually complete within 24 h after noise exposure. It is generally considered that the loss of afferent synapses is irreversible [
25,
34,
35] but, more recently, post-exposure regeneration of afferent synapses has also been reported after intratympanic administration of neurotrophin-3 [
36]. Given that CCG-4986 was administered 48 h after acoustic trauma, it is unclear how it prevents synaptic loss or regenerates afferent synapses. Further studies are required to investigate the timeline of IHC synaptopathic injury in the rat cochlea and establish the underlying mechanism of the rescue effect by CCG-4986.
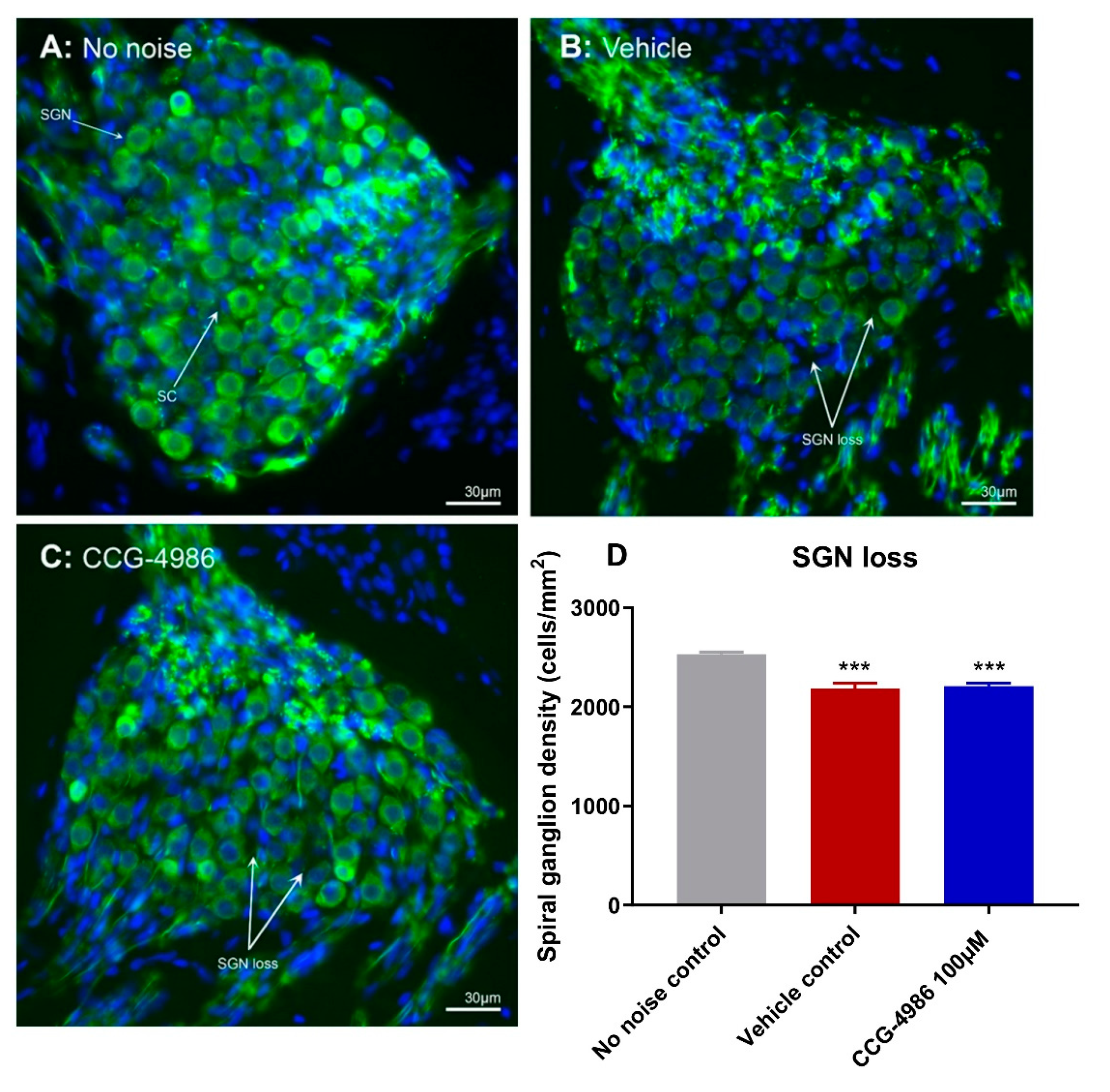
CCG-4986 treatment 48 h post-insult, however, did not protect against SGN loss. SGN loss usually progresses at a much slower rate than the loss of afferent synapses after acoustic overexposure [
25]. Since SGN loss was measured only at one time point (16 days after insult), further studies investigating SGN survival months after acoustic insult are required to fully assess the neuroprotective effect of CCG-4986.
3.5. Putative Mechanisms of Otoprotection by CCG-4986
CCG-4986 is a small molecule inhibitor of RGS4 which enhances adenosine A
1R signalling by disrupting the molecular complex (Neurabin/RGS4) that terminates A
1R signalling [
18]. RGS4 is a GTP-activating protein (GAP) that selectively terminates A
1R signalling at the level of G protein activation, by accelerating the intrinsic GTPase self-mediated hydrolysis and thus reverting Gα and Gβγ subunits to an inactive state [
37]. This effectively terminates the intracellular response following adenosine A
1R activation.
CCG-4986 blocks RGS4 activity by a dual mode of inhibition, through covalent modification of two different surface-exposed cysteine residues. Firstly, CCG-4986 modifies the Cys132 residue near the binding site of Gα protein and competitively prevents Gα protein binding to RGS4, which moderately reduces the binding affinity of Gα protein to RGS4 [
37,
38]. The second, more dominant mechanism, involves modification of the Cys148 residue located in an allosteric site that causes a conformational change in RGS4 that prevents Gα from interacting with RGS4, subsequently inhibiting GAP activity and thus extending adenosine A
1R signalling [
37].
Chen et al. [
18] demonstrated that the scaffolding protein neurabin is required to facilitate the interaction between RGS4 and A
1R in the brain. In the absence of A
1R stimulation, RGS4 is localised to the cytosol. After A
1R activation, RGS4 is recruited by neurabin to the cell surface, forming the A
1R/neurabin/RGS4 complex that specifically regulates A
1R signalling.
Our study shows that CCG-4986 can partially rescue the cochlea from noise-induced injury, most likely by enhancing A
1R signalling. Noise exposure can induce the release of adenosine into the cochlear fluids and lead to up-regulation of A
1R expression in cochlear tissues [
3,
39]. Adenosine A
1R signalling reduces oxidative stress and lipid peroxidation, likely by boosting endogenous antioxidant defences, including superoxide dismutase and glutathione peroxidase activity [
3,
40]. A
1 receptors in the central nervous system are known to exhibit an inhibitory tone by preventing neuronal excitability and synaptic transmission [
41]. A
1R activation thus has a potential to directly counteract the main mechanisms of noise-induced cochlear injury, including oxidative stress, calcium overload, and glutamate excitotoxicity [
23,
33].
There is a certain advantage of CCG-4986 treatment over adenosine A
1R agonists such as ADAC and R-PIA, which activate A
1R with greater selectivity than adenosine. Our previous study [
23] demonstrated a biphasic dose-response relationship effect of ADAC after systemic administration, otoprotective at doses 100–200 μg/kg and less effective at higher doses. This "effect inversion" might be due to overstimulation of A
1R causing their desensitisation and internalisation [
42]. In contrast, inhibition of the neurabin/RGS4 complex bolsters the A
1R signalling without causing a change in A
1R number or affinity [
18].
4. Materials and Methods
4.1. Animals
For this study, male Wistar rats (6–8 weeks) were sourced from the animal facility at the University of Auckland. Animals were housed in standard cages with ad libitum access to food and water, under controlled conditions (constant humidity and temperature, 12-h light/dark cycle). A minimum of two animals were housed together, up to a maximum of four per cage. Animal welfare was continuously assessed during the study to ensure that animal health was maintained at the highest standard. Noise-exposed animals were randomly assigned into drug treatment or drug vehicle-treated (control) group. All experimental procedures were carried out with the approval of the University of Auckland Animal Ethics Committee (approval # 1631, 8 September 2015), in agreement with the Animal Welfare Act (1999).
4.2. Auditory Brainstem Responses
Auditory brainstem responses (ABR) were used to determine auditory thresholds in rats prior to noise exposure (baseline) and 14 days after intratympanic drug or vehicle injection (final). The ABR is an auditory evoked potential that represents the synchronised summation of neuronal activity in response to auditory clicks and tone pip stimuli. ABR recordings were made in a custom-built sound isolating chamber (Shelberg Acoustics, Sydney, Australia), equipped with internal ventilation and a light source. Animals were anesthetised with a mixture of Ketamine (25 mg/kg) and Domitor (0.5 mg/kg) given intraperitoneally. The tympanic membrane was checked for signs of infection, physical trauma, or scarring before ABR recording. Only the left ear was used for assessment of ABR thresholds in each animal. Animals were placed on a thermostatically controlled electric blanket during recordings to maintain body temperature at 37 °C. The Tucker-Davis Technologies (TDT) System 3 and BioSig digital signal processing software (Alachua, FL, USA) were used to generate the auditory stimuli. A multi-field magnetic speaker (MF1, TDT) with 10 cm plastic tubing was used to deliver auditory stimuli into the ear. Three subdermal needle electrodes connected to a Medusa RA4LI headstage amplifier (×20 gain) were used to measure ABR responses. The active electrode was placed at the vertex of the scalp, the reference electrode at the mastoid region of the ear of interest, and the ground electrode at the mastoid region of the contralateral ear. Tone pips (5 ms duration, 2 ms rise/fall, presented at a 21/s rate) were used to elicit ABR responses at intensities between 90 dB SPL to 5 dB SPL, presented in decremental 5 dB steps. Tone pip responses were acquired at an alternating polarity sampling rate of 512 and averaged for each sound intensity. A bandpass filter (300–3000 Hz, 50 Hz notch) was applied to all responses. The ABR wave I was used to determine auditory thresholds defined as the lowest sound intensity level capable of eliciting a reproducible waveform. The cut off amplitude was set at ≥120 nV, as consistently reproducible waveforms were obtained at and above this amplitude. ABR recordings were repeated at sound intensities 10 dB above and 5 dB below the threshold in 5 dB decrements to confirm threshold intensity. In cases where the threshold ceiling was exceeded (above 90 dB SPL), these thresholds were arbitrarily assigned a value of 95 dB SPL. ABR assessments were carried out one day prior to noise exposure (baseline) and 14 days after intratympanic injection (final).
The amplitudes and latencies of wave I were assessed at suprathreshold intensities for selected frequencies (4, 16, and 28 kHz) to investigate the effect of CCG-4986 treatment on noise-induced neuronal injury. Animals with final auditory thresholds of 80 dB SPL or lower were included for input-output functional analysis. The amplitude of wave I (peak to trough) was measured at 90, 85, and 80 dB SPL intensities, and latency was measured as the time taken to reach the peak (including 0.3 ms signal transduction time from the speaker to the ear).
4.3. Noise Exposure
Twenty-four hours after baseline ABR measurements, animals were exposed to an octave band noise (8–16 kHz) for 2 h at 110 dB SPL. Acoustic overstimulation was carried out in a custom-built sound-attenuating chamber (Shelburg Acoustics, Sydney, Australia), equipped with internal ventilation, light source, and speakers suspended from the ceiling. Frequency and intensity of sound were adjusted by external controls. The speakers were calibrated using a sound level meter (Precision Sound level Meter Type 2235, Brüel & Kjær; Nærum, Denmark) prior to each noise exposure session, with the average sound pressure intensity measuring 110 ± 1.5 dB SPL across the cage floor. Animals exposed to noise were placed inside a conventional rat cage, positioned with a 30 cm distance underneath the speakers, and allowed to acclimatise for 5 min. Sound intensity was gradually increased over a period of 5 min. Control animals were placed in the sound isolating chamber for two hours to control for relocation stress. Afterwards, animals were returned to the animal housing facility and kept at ambient noise levels (55–65 dB SPL) for the remainder of the experimental timeline.
4.4. Intratympanic Injections
RGS4 inhibitor CCG-4986 or drug vehicle solution (control) was delivered by intratympanic injection into the middle ear cavity 24 or 48 h after noise exposure. Drug solution was made by dissolving CCG-4986 (ChemBridge™; San Diego, CA, USA) in 1% DMSO and 0.9% saline with the final 100 μM working dilution for intratympanic injection. Control animals were treated with the drug vehicle solution (1% DMSO in 0.9% saline). Drug and vehicle solutions were mixed with 17%
w/w poloxamer-407 (Sigma-Aldrich) and placed on ice until fully dissolved. Solutions were then aliquoted and stored at −20 °C for later use. Poloxamer-407 allows for slow drug delivery to the cochlea as the solution becomes a gel at body temperature [
21].
Prior to drug treatment, animals were anaesthetised with a mixture of Ketamine (25 mg/kg) and Domitor (0.5 mg/kg) injected intraperitoneally and administered one dose of Temgesic (Buprenorphine; 0.05 mg/kg, subcutaneously) for analgesia. Intratympanic injections were carried out using a Hamilton syringe mounted on a micromanipulator arm. The needle was inserted through the posterior-superior quadrant of the tympanic membrane to deliver injection solution into the vicinity of the round window membrane of the cochlea. A total solution volume of 27 μL was slowly injected into the middle ear. The animal was then returned to a recovery cage and left on its side for 30 min to allow the solution to settle onto the RWM in a gel form. Then the procedure was repeated for the contralateral ear. Animals were then given a subcutaneous dose of Antisedan (1 mg/kg) to reverse the effects of ketamine/domitor anaesthesia.
4.5. Cochlear Tissue Preparation for Histology and Immunohistochemistry
After the final ABR assessment, animals were euthanized by an anaesthetic overdose (pentobarbitone, 90–100 mg/kg intraperitoneally). The animals were perfused with the flush solution (0.9% NaCl containing 10% NaNO2) and then tissue fixative (4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB, pH 7.4) overnight at 4 °C. Cochleae were then decalcified using 5% EDTA in 0.1 M PB for 9 days at 4 °C, cryoprotected overnight in 30% sucrose in 0.1 M PB and embedded in optimal cutting temperature compound (OCT). Cochleae were then snap frozen using N-pentane and stored at −80 °C for further processing.
4.6. Hair Cell and Ribbon Synapse Counting
The extracted cochleae were decapsulated and micro-dissected into the apical, middle, and basal segments (turns), after removal of the lateral wall, Reissner’s membrane and tectorial membrane. Cochlear turns were transferred into a 48 well plate containing 0.1 M PBS (pH 7.4), permeabilised with 1% Triton X-100 and blocked with 10% Normal Goat Serum (NGS) for 2 h at room temperature (RT). Whole mount tissues were then incubated overnight at RT with the following primary antibodies: rabbit polyclonal anti-Myosin-VIIa (Proteus Biosciences, 1:500), mouse anti-C-terminal binding protein 2 (CtBP2; IgG1; BD Biosciences, 1:500) and mouse anti-Glutamate receptor 2 (GluA2; IgG2; Merck Millipore, 1:500) in antibody solution containing 0.1% Triton X-100 in 0.1 M PBS with 5% NGS. The following day, sections were washed three times for 60 min with 0.1 M PBS and then incubated at RT with the following secondary antibodies: goat anti-rabbit (Alexa 568, 1:500; Invitrogen), goat anti-mouse IgG1 (Alexa 647, 1:500; Invitrogen), and goat anti-mouse IgG2 (Alexa 488, 1:500; Invitrogen) in antibody solution containing 0.1% Triton X-100 and 5% NGS in 0.1 M PBS. Whole mounts were then washed for 60 min with 0.1 M PBS and then mounted on glass slides using Citifluor AFI mounting medium, cover slipped, sealed with nail polish, and stored in the dark at 4 °C. Inner hair cell-auditory nerve synapses were imaged and analysed at three frequency regions (4, 16, and 28 kHz) of the cochlea, based on the distance from the cochlear apex [
43]. Immunostained synapses were imaged using confocal microscopy (Olympus FV1000 Live Cell System, Tokyo, Japan) with oil immersion 60× objective (1.35 NA) and 2.6× digital zoom. Images were captured as a z-stack, with the
z dimension sampled in 0.2 μm steps, imaging frame in the
xy dimension capturing 10 adjacent inner hair cells. Z-stacks were processed to remove nuclear CtBP2 staining and compiled into a colour composite stack: inner hair cells labelled with Myosin-VIIa (grey), post-synaptic terminals labelled with GluA2 (green), pre-synaptic ribbons labelled with CtBP2 (red). As a max z-projection could potentially obscure juxtaposed paired synapses in the z-dimension, synapses were counted frame by frame in the z-dimension using the cell counter plugin in ImageJ, with markers displayed through the stack to avoid duplicate counts. A paired synapse was defined as a post-synaptic terminal (GluA2) immediately adjacent to a presynaptic ribbon (CtBP2). Any unpaired pre-synaptic ribbons or post-synaptic terminals were defined as orphan synapses. The total number of synapses were counted for 10 inner hair cells and averaged to get the mean number of synapses per IHC. The number of unpaired (orphan) synapses was expressed as a percentage of the total synapse count per IHC.
For hair cell counting, each cochlea was divided into three segments covering the entire length of the cochlea and representing different frequency regions. The apical segment occupied approximately 0–30% from the apex, middle segment 30–75% from the apex, and basal segment 75–100% from the apex. The organ of Corti was imaged with a Zeiss Axioplan 2 epifluorescence microscope (Carl Zeiss, Jena, Germany) with 20× objective (0.6 NA), and captured with a Photometrics Prime sCMOS monochrome camera. Inner and outer hair cells were counted in ImageJ using the CellCounter to mark intact and missing hair cells, with the number of missing cells expressed as a percentage of the total number of hair cells counted. For regions with complete OHC loss, but intact IHC, one IHC was approximated to three missing OHC (one in each row). For regions of absolute hair cell loss, an adjacent length of IHC was measured (3–4 cells) to calculate pixel width per IHC. The distance of the lesion was measured and the number of missing IHC and corresponding missing OHC was estimated.
4.7. Spiral Ganglion Neuron Counting
Cochleae designated for spiral ganglion neuron counts were cryosectioned at 12 μm, and every second mid-modiolar section was placed into 0.1 M PBS (pH 7.4) in a 24 well plate. Cryosections were washed with 0.1 M PBS, permeabilised with 1% Triton X-100 in 0.1 M PBS and blocked with 10% normal donkey serum (NDS) for 1 h at RT. Cochlear sections were then incubated overnight at 4 °C with goat polyclonal Neurofilament (NF-L) antibody (2 μg/mL; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) in antibody solution containing 0.1% Triton X-100 and 10% NDS in 0.1 M PBS. The next day sections were washed (3 × 10 min) with 0.1 M PBS and incubated with donkey anti-goat secondary antibody (Alexa Fluor 488, 1:600 dilution; Invitrogen) for 2 h at RT. Sections were then washed with 0.1 M PBS (10 min), incubated with Hoechst 33342 nuclear stain (1 μg/mL in 0.1 M PBS, pH 7.4; Thermo Fisher Scientific) for 15 min, then washed with 0.1 M PBS for 10 min. Sections were mounted on a glass slide in Citifluor AF1 mounting medium, covered with a coverslip and stored in the dark at 4 °C. Tissues were imaged with a Zeiss Axioplan 2 epifluorescence microscope (Carl Zeiss, Jena, Germany) with 40× objective. Images of the spiral ganglion neurons located in the middle turn were captured at two different wavelengths (UV and 488) using a Photometrics Prime sCMOS monochrome camera and merged to identify individual spiral ganglion neurons. The spiral ganglion area, represented by the bony edge of Rosenthal’s canal, was selected using the "free-hand selection tool" in ImageJ. Individual neurons with unambiguous round nuclei in the middle turn of the cochlea were counted in each section and then averaged to determine SGN density for each animal as described previously [
44].
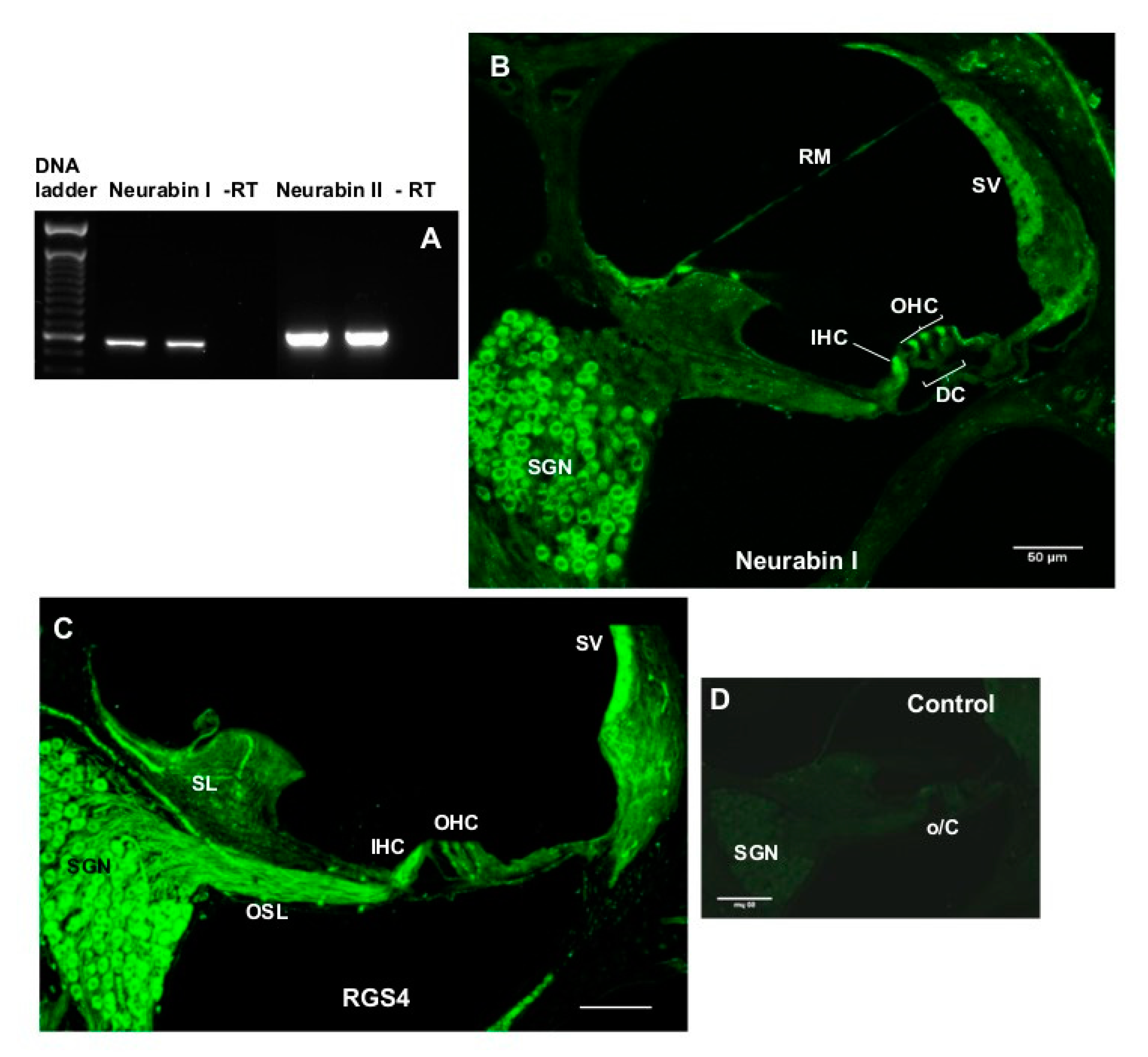
4.8. Characterisation of Neurabin Expression in the Rat Cochlea
To determine mRNA expression of the two neurabin isoforms (Neurabin I and II), four intact rat cochleae were extracted, decapsulated, and placed into separate Eppendorf tubes containing cold lysis buffer (100 mM TRIS-HCl pH 7.5, 500 mM LiCl, 10 mM EDTA pH 8.0, 1% LiDS, 5 mM dithiothreitol (DTT) and RNase inhibitors) pre-chilled to 4 °C. Cochleae were then homogenised using sterile Teflon mini-pestles. Polyadenylated RNA (mRNA) was extracted using the magnetic Dynabeads
® (Oligo(dT)25 (5 mg/mL)) mRNA DIRECT kit (Invitrogen). First-strand cDNA synthesis was carried out in a 20-μl reverse transcription (RT) reaction with random hexamers, dNTPs and Superscript III reverse transcriptase (Invitrogen). The complementary DNA was amplified by PCR with rat-specific primers for neurabin isoforms (
Table 1) designed using OligoPerfect™ (Invitrogen). Negative control without reverse transcriptase was included in each PCR run. RT-PCR with a 40 cycle profile was performed as follows: 94 °C denaturation (1 min), 60 °C annealing (1.5 min), 72 °C extension (2 min) steps using PTC-100
™Programmable Thermal Controller (MJ Research Inc., Waltham, MA, USA). PCR amplicons were separated by agarose gel electrophoresis, and visualised using SYBR safe DNA gel stain (Invitrogen). PCR products were purified by PureLink™ PCR Purification Kit (Invitrogen) and the identity of the amplicons confirmed by DNA sequencing (Centre for Genomics & Proteomics, School of Biological Sciences, the University of Auckland, Auckland, New Zealand).
4.9. RGS4 and Neurabin Immunohistochemistry
The immunolocalisation of RGS4 and neurabin I isoform which forms molecular complexes with RGS4 was demonstrated in the rat cochleae using immunofluorescence. Briefly, rat cochlear tissues were cryosectioned at 30 μm using a CM3050 S cryostat (Leica, Germany) and mid-modiolar sections were placed in a 24 well plate containing 0.1 M PBS (pH 7.4) and washed twice for 20 min. Sections were then incubated in blocking and permeabilisation solution (10% NGS, 1% Triton X-100 in 0.1 M PBS) for 1 h at RT, followed by incubation with a mouse monoclonal RGS4 antibody (Santa Cruz Biotechnologies, sc-398658; 2 μg/mL) or rabbit polyclonal neurabin I antibody (Santa Cruz Biotechnologies; 2 μg/mL) overnight at 4 °C. Control sections were incubated with the normal mouse or rabbit IgG (2 μg/mL) instead of the primary antibody. The next day, sections were washed with 0.1 M PBS three times (60 min), followed by the incubation with the secondary antibody (Alexa Fluor 488 goat anti-mouse or anti-rabbit IgG, dilution 1:500) for 2 h at RT. After a washout with 0.1 M PBS (3 × 10 min), cryosections were mounted on glass slides using Citifluor AF1 Mounting Medium, covered with a coverslip and sealed with nail polish. Images of mid-modiolar cochlear cryosections were acquired using a confocal microscope (Olympus FluoView FV1000) and processed with FluoView software (version 2.0c, Olympus, Tokyo, Japan).
4.10. Data Analysis
The researchers were blinded for all ABR assessments, tissue collections, and histological analyses. Animals were assigned a subject ID by an independent researcher and allocated into the treatment or vehicle control group using a randomly generated number list (
https://www.randomizer.org). Aliquoted injection solutions were labelled only by subject ID. All data were tested for normality using the Shapiro–Wilk Test. Auditory thresholds, ribbon synapse and hair cell counts were analysed using a two-way ANOVA with a post-hoc Holm–Sidak test. Spiral ganglion counts were analysed with one-way ANOVA. Suprathreshold data were analysed using multi-level factorial ANOVA with planned orthogonal contrasts to determine differences between groups. Data are presented as mean ± SEM.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}