Identification of Key Receptor Residues Discriminating Human Chorionic Gonadotropin (hCG)- and Luteinizing Hormone (LH)-Specific Signaling


 and
and
Abstract
1. Introduction
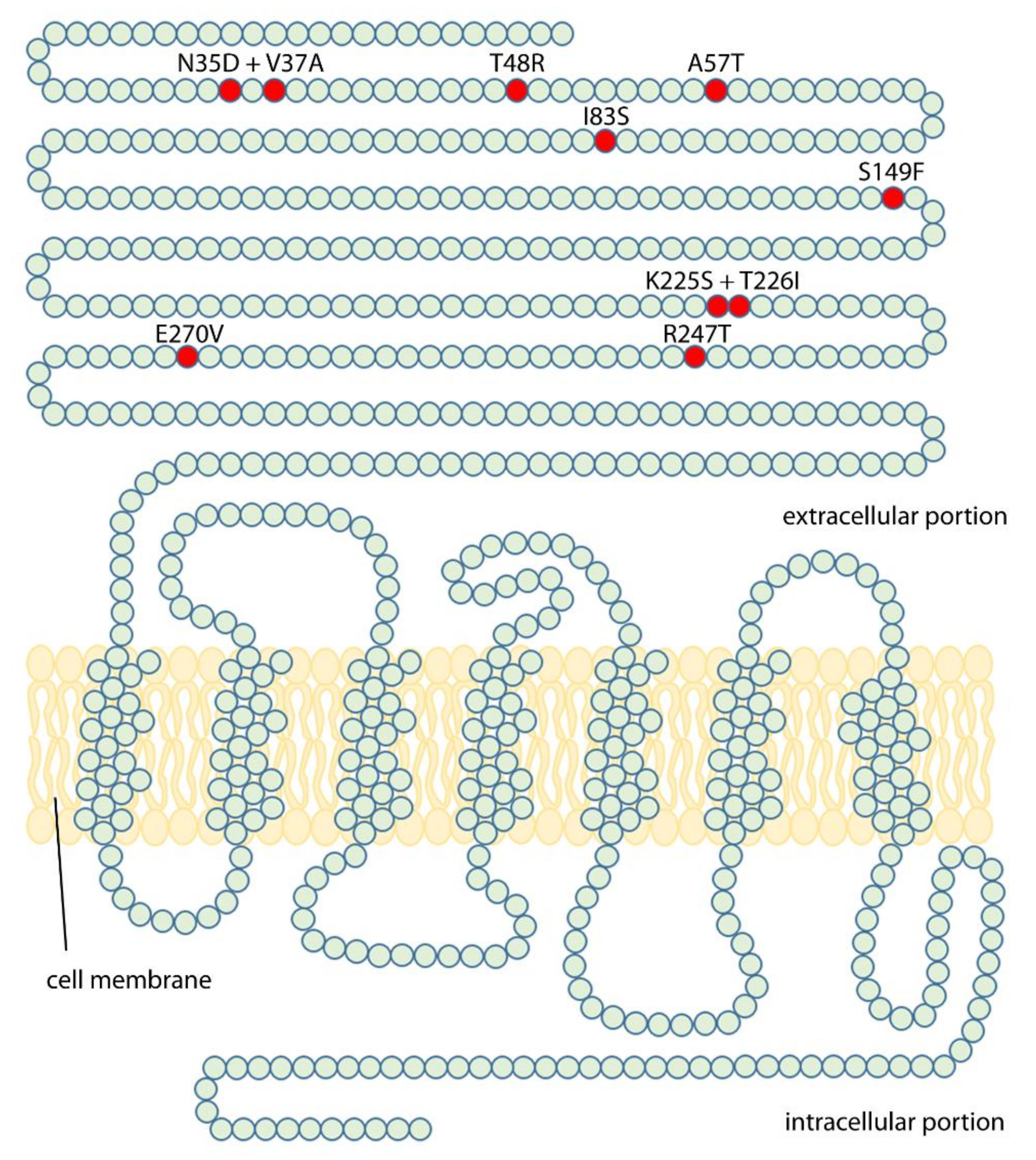
2. Results
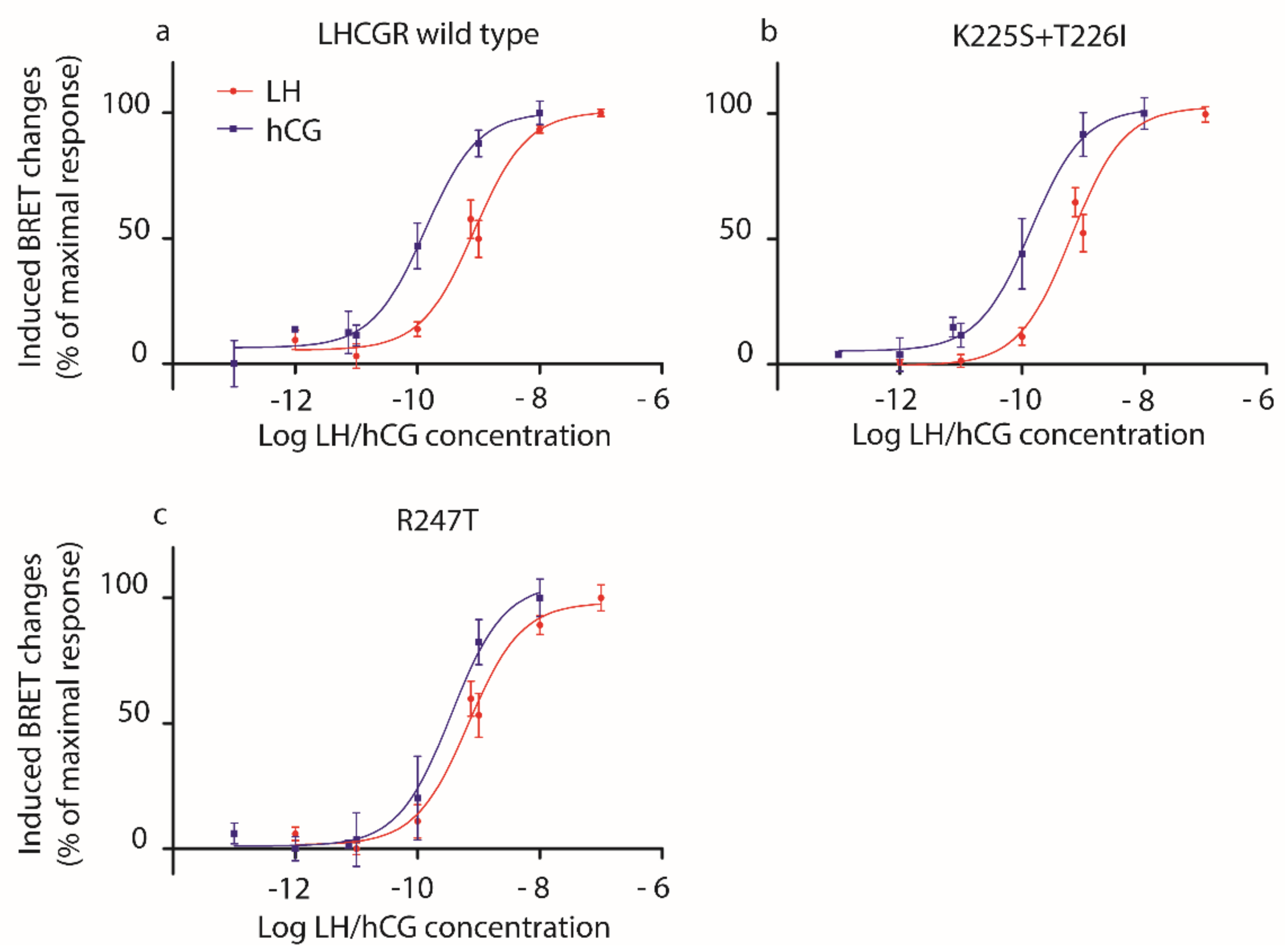
2.1. Analysis of LH- and hCG-Induced cAMP Production
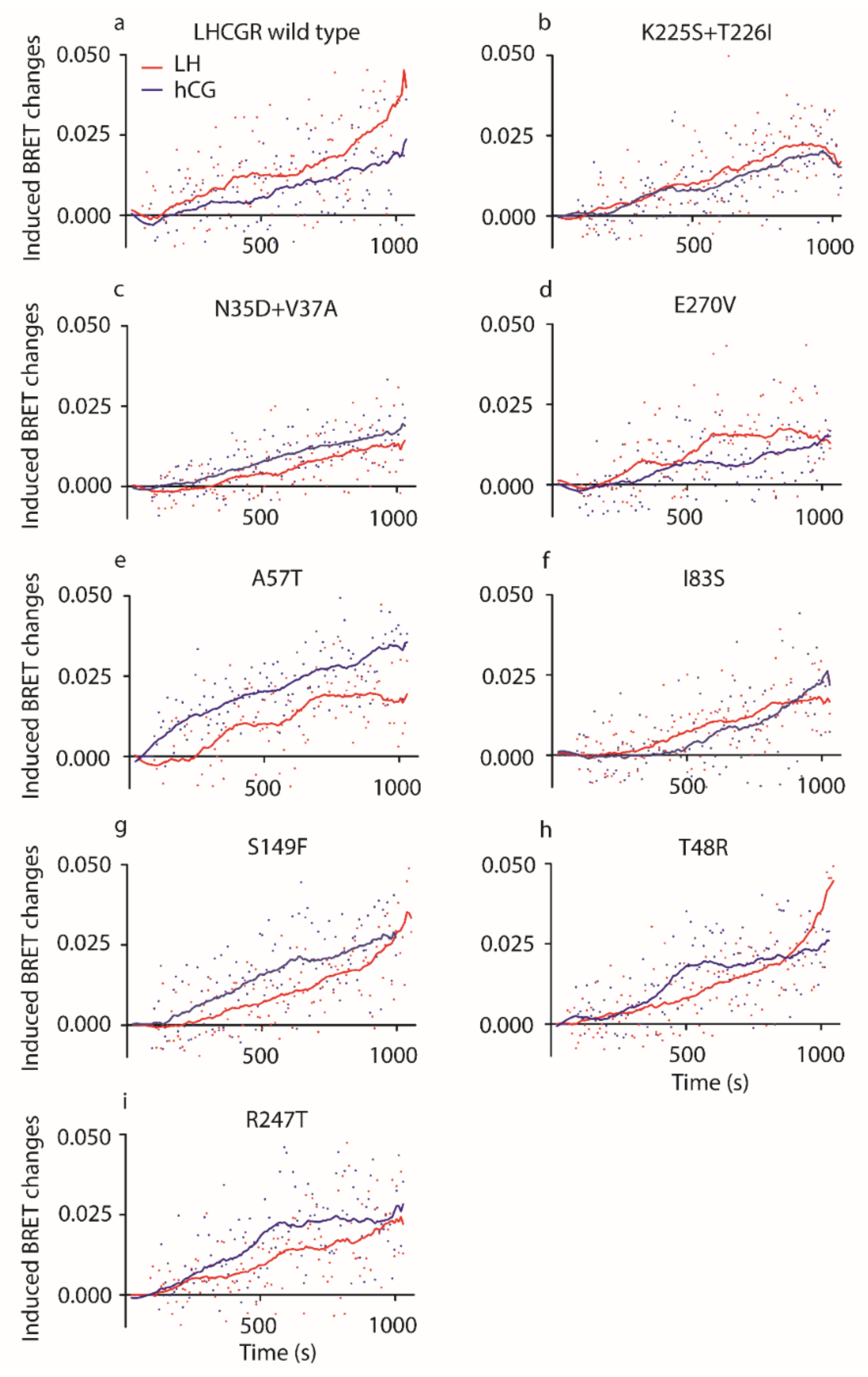
2.2. Analysis of LH- and hCG-Induced ERK1/2 Phosphorylation
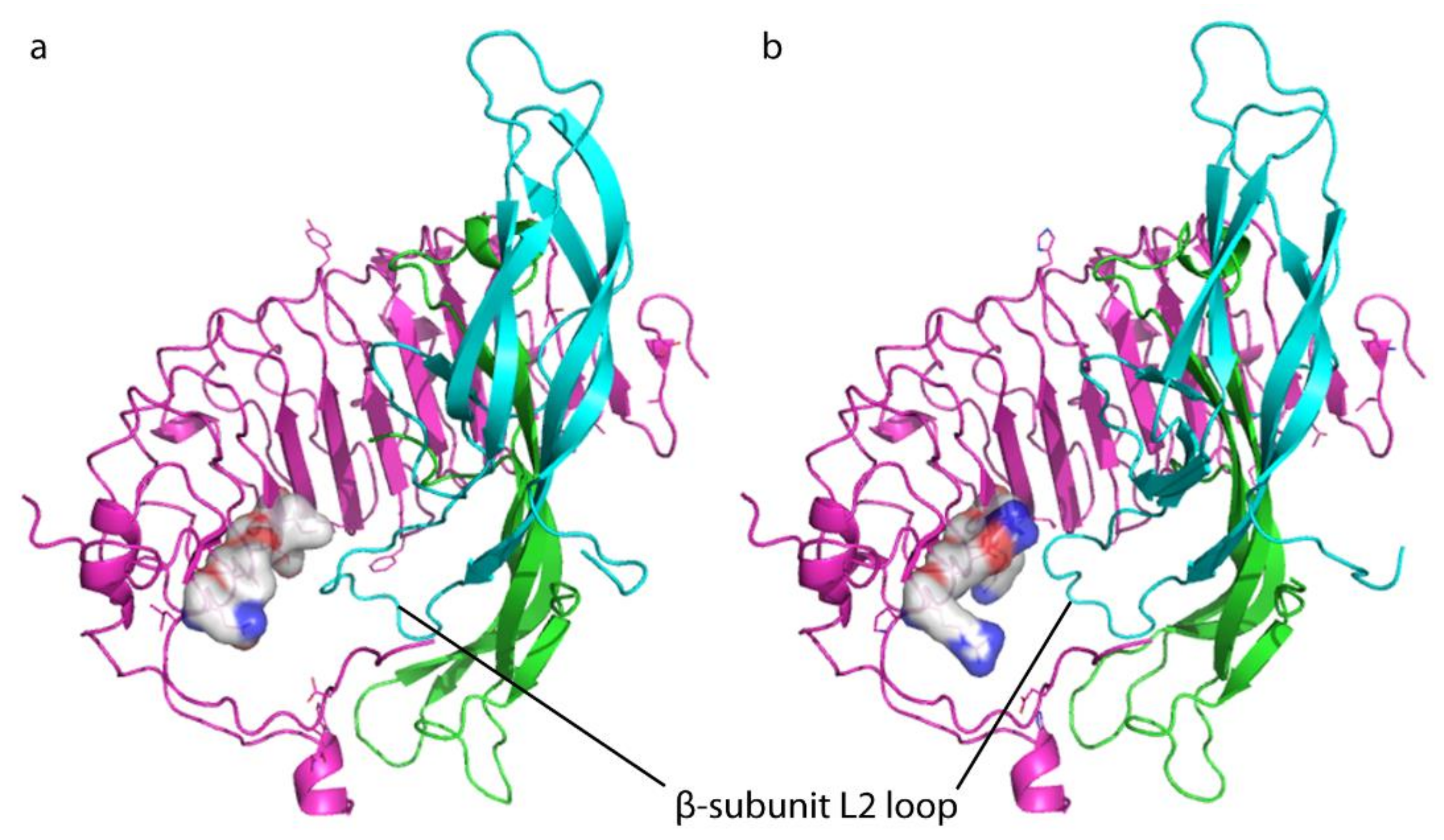
3. Discussion
4. Materials and Methods
4.1. Recombinant Hormones
4.2. HEK293 Cell Line Cultures and Handling
4.3. Plasmid Vectors of Mutant and Wild Type LHCGR
4.4. Transfection Protocols
4.5. cAMP Measurement by BRET
4.6. Evaluation of pERK1/2 Activation by BRET
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LH. | Luteinizing hormone |
| hCG | Human chorionic gonadotropin |
| GPCR | G protein-coupled receptor |
| LHCGR | Human LH/hCG receptor |
| Lhr | Murine luteinizing-hormone receptor |
| CTP | C-terminal extension |
| LRRD | Leucine-rich-repeat domain |
| cAMP | Cyclic adenosine monophosphate |
| PKA | Protein kinase A |
| AKT | Protein kinase B |
| ERK1/2 | Extracellular signal-regulated kinases 1/2 |
| HEK293 | Human Embryonic kidney 293 |
| DMEM | Dulbecco’s Modified Eagles Medium |
| FBS | fetal bovine serum |
| EDTA | Ethylenediaminetetraacetic acid |
| PBS | Phosphate buffered saline |
| BRET | Bioluminescence resonance energy transfer |
| pERK1/2 | Phosphorylated extracellular signal-regulated kinases 1/2 |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| IBMX | Phosphodiesterase inhibitor 3-isobutil-1-methylxanthine |
| EC50 | 50% effective concentrations |
| LOWESS | Locally weighted scatterplot smoothing |
| AUC | Area under the curve |
| SEM | Standard error of the mean |
| PMA | Phorbol 12-myristate 13-acetate |
References
- Ascoli, M.; Fanelli, F.; Segaloff, D.L. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr. Rev. 2002, 23, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Santi, D.; Brigante, G.; Simoni, M. Two hormones for one receptor: Evolution, biochemistry, actions, and pathophysiology of LH and hCG. Endocr. Rev. 2018, 39, 549–592. [Google Scholar] [CrossRef] [PubMed]
- Grzesik, P.; Kreuchwig, A.; Rutz, C.; Furkert, J.; Wiesner, B.; Schuelein, R.; Kleinau, G.; Gromoll, J.; Krause, G. Differences in Signal Activation by LH and hCG are Mediated by the LH/CG Receptor’s Extracellular Hinge Region. Front. Endocrinol. 2015, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Lapthorn, A.J.; Harris, D.C.; Littlejohn, A.; Lustbader, J.W.; Canfield, R.E.; Machin, K.J.; Morgan, F.J.; Isaacs, N.W. Crystal structure of human chorionic gonadotropin. Nature 1994, 369, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Gabay, R.; Rozen, S.; Samokovlisky, A.; Amor, Y.; Rosenfeld, R.; Kohen, F.; Amsterdam, A.; Berger, P.; Ben-Menahem, D. The role of the 3′ region of mammalian gonadotropin β subunit gene in the luteinizing hormone to chorionic gonadotropin evolution. Mol. Cell Endocrinol. 2014, 382, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Wide, L.; Eriksson, K.; Sluss, P.M.; Hall, J.E. Serum half-life of pituitary gonadotropins is decreased by sulfonation and increased by sialylation in women. J. Clin. Endocrinol. Metab. 2009, 94, 958–964. [Google Scholar] [CrossRef]
- Jiang, X.; Dias, J.A.; He, X. Structural biology of glycoprotein hormones and their receptors: Insights to signaling. Mol. Cell Endocrinol. 2014, 382, 424–451. [Google Scholar] [CrossRef]
- Casarini, L.; Santi, D.; Simoni, M.; Potì, F. ‘spare’ Luteinizing Hormone Receptors: Facts and Fiction. Trends Endocrinol. Metab. 2018, 29, 208–217. [Google Scholar] [CrossRef]
- Riccetti, L.; De Pascali, F.; Gilioli, L.; Potì, F.; Giva, L.B.; Marino, M.; Tagliavini, S.; Trenti, T.; Fanelli, F.; Mezzullo, M.; et al. Human LH and hCG stimulate differently the early signalling pathways but result in equal testosterone synthesis in mouse Leydig cells in vitro. Reprod. Biol. Endocrinol. 2017, 15, 2. [Google Scholar] [CrossRef]
- Casarini, L.; Lispi, M.; Longobardi, S.; Milosa, F.; la Marca, A.; Tagliasacchi, D.; Pignatti, E.; Simoni, M. LH and hCG Action on the Same Receptor Results in Quantitatively and Qualitatively Different Intracellular Signalling. PLoS ONE 2012, 7, e46682. [Google Scholar] [CrossRef]
- Huhtaniemi, I.T.; Catt, K.J. Differential binding affinities of rat testis luteinizing hormone (lH) receptors for human chorionic gonadotropin, human lH, and ovine LH. Endocrinology 1981, 108, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Hallast, P.; Nagirnaja, L.; Margus, T.; Laan, M. Segmental duplications and gene conversion: Human luteinizing hormone/chorionic gonadotropin β gene cluster. Genome Res. 2005, 15, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Nagirnaja, L.; Rull, K.; Uusküla, L.; Hallast, P.; Grigorova, M.; Laan, M. Genomics and genetics of gonadotropin beta-subunit genes: Unique FSHB and duplicated LHB/CGB loci. Mol. Cell Endocrinol. 2010, 329, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Galet, C.; Ascoli, M. The differential binding affinities of the luteinizing hormone (LH)/choriogonadotropin receptor for LH and choriogonadotropin are dictated by different extracellular domain residues. Mol. Endocrinol. 2005, 19, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Riccetti, L.; Yvinec, R.; Klett, D.; Gallay, N.; Combarnous, Y.; Reiter, E.; Simoni, M.; Casarini, L.; Ayoub, M.A. Human Luteinizing Hormone and Chorionic Gonadotropin Display Biased Agonism at the LH and LH/CG Receptors. Sci. Rep. 2017, 7, 940. [Google Scholar] [CrossRef] [PubMed]
- Grzesik, P.; Teichmann, A.; Furkert, J.; Rutz, C.; Wiesner, B.; Kleinau, G.; Schülein, R.; Gromoll, J.; Krause, G. Differences between lutropin-mediated and choriogonadotropin-mediated receptor activation. FEBS J. 2014, 281, 1479–1492. [Google Scholar] [CrossRef]
- Müller, T.; Gromoll, J.; Simoni, M. Absence of exon 10 of the human luteinizing hormone (LH) receptor impairs LH, but not human chorionic gonadotropin action. J. Clin. Endocrinol. Metab. 2003, 88, 2242–2249. [Google Scholar] [CrossRef]
- Gromoll, J.; Eiholzer, U.; Nieschlag, E.; Simoni, M. Male hypogonadism caused by homozygous deletion of exon 10 of the luteinizing hormone (LH) receptor: Differential action of human chorionic gonadotropin and LH. J. Clin. Endocrinol. Metab. 2000, 85, 2281–2286. [Google Scholar] [CrossRef]
- Casarini, L.; Reiter, E.; Simoni, M. β-arrestins regulate gonadotropin receptor-mediated cell proliferation and apoptosis by controlling different FSHR or LHCGR intracellular signaling in the hGL5 cell line. Mol. Cell. Endocrinol. 2016, 437, 11–21. [Google Scholar] [CrossRef]
- Tranchant, T.; Durand, G.; Gauthier, C.; Crépieux, P.; Ulloa-Aguirre, A.; Royère, D.; Reiter, E. Preferential β-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol. Cell. Endocrinol. 2011, 331, 109–118. [Google Scholar] [CrossRef]
- Casarini, L.; Lazzaretti, C.; Paradiso, E.; Limoncella, S.; Riccetti, L.; Sperduti, S.; Melli, B.; Marcozzi, S.; Anzivino, C.; Sayers, N.S.; et al. Membrane estrogen receptor (GPER) and follicle-stimulating hormone Receptor (FSHR) Heteromeric complexes promote human ovarian follicle survival. iScience 2020, 23, 101812. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.C.; Fanelli, F.; Huhtaniemi, I.T.; Hanyaloglu, A.C. Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J. Biol. Chem. 2015, 290, 3875–3892. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Riccetti, L.; de Pascali, F.; Gilioli, L.; Marino, M.; Vecchi, E.; Morini, D.; Nicoli, A.; La Sala, G.B.; Simoni, M. Estrogen modulates specific life and death signals induced by LH and hCG in human primary granulosa cells in vitro. Int. J. Mol. Sci. 2017, 18, 926. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.; Casarini, L.; Alviggi, C.; Simoni, M. Efficacy of follicle-stimulating hormone (FSH) alone, FSH + luteinizing hormone, human menopausal gonadotropin or FSH + human chorionic gonadotropin on assisted reproductive technology outcomes in the “personalized” medicine era: A meta-analysis. Front. Endocrinol. 2017, 8, 114. [Google Scholar] [CrossRef]
- Van Loy, T.; Vandersmissen, H.P.; Van Hiel, M.B.; Poels, J.; Verlinden, H.; Badisco, L.; Vassart, G.; Vanden Broeck, J. Comparative genomics of leucine-rich repeats containing G protein-coupled receptors and their ligands. Gen. Comp. Endocrinol. 2008, 155, 14–21. [Google Scholar] [CrossRef]
- Moyle, W.R.; Lin, W.; Myers, R.V.; Cao, D.; Kerrigan, J.E.; Bernard, M.P. Models of glycoprotein hormone receptor interaction. Endocrine 2005, 26, 189–205. [Google Scholar] [CrossRef]
- Campbell, R.K.; Satoh, N.; Degnan, B.M. Piecing together evolution of the vertebrate endocrine system. Trends Genet. 2004, 20, 359–366. [Google Scholar] [CrossRef]
- Cole, L.A.; Khanlian, S.A.; Kohorn, E.I. Evolution of the human brain, chorionic gonadotropin and hemochorial implantation of the placenta: Insights into origins of pregnancy failures, preeclampsia and choriocarcinoma. J. Reprod. Med. 2008, 53, 549–557. [Google Scholar]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef]
- Xu, C.; Peter, M.; Bouquier, N.; Ollendorff, V.; Villamil, I.; Liu, J.; Fagni, L.; Perroy, J. REV, a BRET-based sensor of ERK activity. Front. Endocrinol. 2013, 4, 95. [Google Scholar] [CrossRef]
- Metzger, H.; Lindner, E. The positive inotropic-acting forskolin, a potent adenylatecyclase activator. Arzneim. Forsch. Drug Res. 1981, 31, 1248–1250. [Google Scholar]
- Lazzaretti, C.; Riccetti, L.; Sperduti, S.; Anzivino, C.; Brigante, G.; De Pascali, F.; Potì, F.; Rovei, V.; Restagno, G.; Mari, C.; et al. Inferring biallelism of two FSH receptor mutations associated with spontaneous ovarian hyperstimulation syndrome by evaluating FSH, LH and HCG cross-activity. Reprod. Biomed. Online 2019, 38, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Moriondo, V.; Marino, M.; Adversi, F.; Capodanno, F.; Grisolia, C.; La Marca, A.; La Sala, G.B.; Simoni, M. FSHR polymorphism p.N680S mediates different responses to FSH in vitro. Mol. Cell. Endocrinol. 2014, 393, 83–91. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor. | Cell Type | Hormone | cAMP EC50 | pERK1/2 ECmax | cAMP:pERK1/2 Ratio | Ref. |
|---|---|---|---|---|---|---|
| LHCGR | Human Granulosa | LH | 530 ± 51 | 100 | 5:1 | [10] |
| hCG | 107 ± 14 | 100 | 1:1 | |||
| Lhr | Mouse Leydig | LH | 192 ± 54 | 100 | 2:1 | [9] |
| hCG | 18 ± 10 | 10 | 2:1 |
| LHCGR Mutation | LH EC50 (pM) | hCG EC50 (pM) | p Value |
|---|---|---|---|
| K225S + T226I | 697.5 ± 165.9 | 193.5 ± 100.2 | 0.0571 |
| N35D + V37A | 595.4 ± 154.8 | 94.49 ± 3.38 | 0.0286 |
| E270V | 2806.0 ± 1393.0 | 248.4 ± 40.65 | 0.0286 |
| A57T | 702.6 ± 222.9 | 152.8 ± 33.91 | 0.0286 |
| I83S | 711.9 ± 187.7 | 127.2 ± 6.28 | 0.0286 |
| S149F | 755.8 ± 173.3 | 111.0 ± 23.66 | 0.0286 |
| T48R | 1207.0 ± 389.1 | 130.5 ± 19.58 | 0.0286 |
| R247T | 556.5 ± 31.63 | 386.2 ± 132.0 | 0.6286 |
| Wild type | 1111.0 ± 378.2 | 142.8 ± 30.85 | 0.0286 |
| LHCGR Mutation | LH AUC | hCG AUC | p Value (LH vs. hCG) | p Value (vs. Wild Type) |
|---|---|---|---|---|
| K225S + T226I | 11.40 ± 1.30 | 9.47 ± 1.70 | ns | ns |
| N35D + V37A | 5.48 ± 0.63 | 8.19 ± 1.35 | ns | <0.0001 |
| E270V | 10.93 ± 1.14 | 7.18 ± 0.93 | ns | ns |
| A57T | 10.89 ± 3.19 | 20.50 ± 4.86 | <0.0001 | <0.0001 |
| I83S | 8.03 ± 1.85 | 6.65 ± 1.08 | ns | <0.0001 |
| S149F | 10.40 ± 1.64 | 13.91 ± 3.07 | ns | ns |
| T48R | 12.28 ± 1.56 | 13.44 ± 3.94 | ns | ns |
| R247T | 10.53 ± 1.07 | 15.44 ± 2.36 | <0.01 | ns |
| Wild type | 13.92 ± 1.60 | 7.98 ± 0.80 | <0.0001 | NA |
| LHCGR Mutation. | LH:hCG EC50 Ratio for cAMP Activation | LH:hCG AUC Ratio for pERK1/2 Activation | Reference |
|---|---|---|---|
| K225S + T226I | 3.6 | 1.2 | Present study |
| N35D + V37A | 6.3 | 0.7 | Present study |
| E270V | 11.3 | 1.5 | Present study |
| A57T | 4.6 | 0.5 | Present study |
| I83S | 5.6 | 1.2 | Present study |
| S149F | 6.8 | 0.7 | Present study |
| T48R | 9.2 | 0.9 | Present study |
| R247T | 1.4 | 0.7 | Present study |
| Wild type | 7.8 | 1.7 | Present study |
| Wild type | 5.0 | 1.0 (calculated on ECmax) | [10] |
| Lhr | 10.7 | 10.0 (calculated on ECmax) | [9] |
| Amino Acid Change | Forward Primer Sequence | Reverse Primer Sequence | Structural Localization |
|---|---|---|---|
| K225S + T226I | CCACAGGGCCGAGTATCTTGGATATTTC | GAAATATCCAAGATACTCGGCCCTGTGG | LRR-9 |
| N35D + V37A | GAGCCCTGCGACTGCGCGCCCGACGGCG | CGCCGTCGGGCGCGCAGTCGCAGGGCTC | Cystein-rich region 1 |
| E270V | CAATCTCCTGGTGGCCACGTTGAC | GTCAACGTGGCCACCAGGAGATTG | Hinge region (LRR-11) |
| A57T | GACTATCACTTACCTACCTCCCTG | CAGGGAGGTAGGTAAGTGATAGTC | LRR-2 |
| I83S | GAAATCTCTCAGAGTGATTCCCTGG | CCAGGGAATCACTCTGAGAGATTTC | LRR-3 |
| S149F | CCTCTGAATTCAATTTCATTCTGG | CCAGAATGAAATTGAATTCAGAGG | LRR-5 |
| T48R | GCCCCGGCCCCAGGGCCGGTCTC | GAGACCGGCCCTGGGGCCGGGGC | Cystein-rich region 1 |
| R247T | GTCCATTCAGACGCTAATTGCCACG | CGTGGCAATTAGCGTCTGAATGGAC | LRR-10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzaretti, C.; Secco, V.; Paradiso, E.; Sperduti, S.; Rutz, C.; Kreuchwig, A.; Krause, G.; Simoni, M.; Casarini, L. Identification of Key Receptor Residues Discriminating Human Chorionic Gonadotropin (hCG)- and Luteinizing Hormone (LH)-Specific Signaling. Int. J. Mol. Sci. 2021, 22, 151. https://doi.org/10.3390/ijms22010151
Lazzaretti C, Secco V, Paradiso E, Sperduti S, Rutz C, Kreuchwig A, Krause G, Simoni M, Casarini L. Identification of Key Receptor Residues Discriminating Human Chorionic Gonadotropin (hCG)- and Luteinizing Hormone (LH)-Specific Signaling. International Journal of Molecular Sciences. 2021; 22(1):151. https://doi.org/10.3390/ijms22010151
Chicago/Turabian StyleLazzaretti, Clara, Valentina Secco, Elia Paradiso, Samantha Sperduti, Claudia Rutz, Annika Kreuchwig, Gerd Krause, Manuela Simoni, and Livio Casarini. 2021. "Identification of Key Receptor Residues Discriminating Human Chorionic Gonadotropin (hCG)- and Luteinizing Hormone (LH)-Specific Signaling" International Journal of Molecular Sciences 22, no. 1: 151. https://doi.org/10.3390/ijms22010151
APA StyleLazzaretti, C., Secco, V., Paradiso, E., Sperduti, S., Rutz, C., Kreuchwig, A., Krause, G., Simoni, M., & Casarini, L. (2021). Identification of Key Receptor Residues Discriminating Human Chorionic Gonadotropin (hCG)- and Luteinizing Hormone (LH)-Specific Signaling. International Journal of Molecular Sciences, 22(1), 151. https://doi.org/10.3390/ijms22010151