The Effect of Chronic Mild Stress and Escitalopram on the Expression and Methylation Levels of Genes Involved in the Oxidative and Nitrosative Stresses as Well as Tryptophan Catabolites Pathway in the Blood and Brain Structures

 , ,
, ,
Abstract
1. Introduction
2. Results
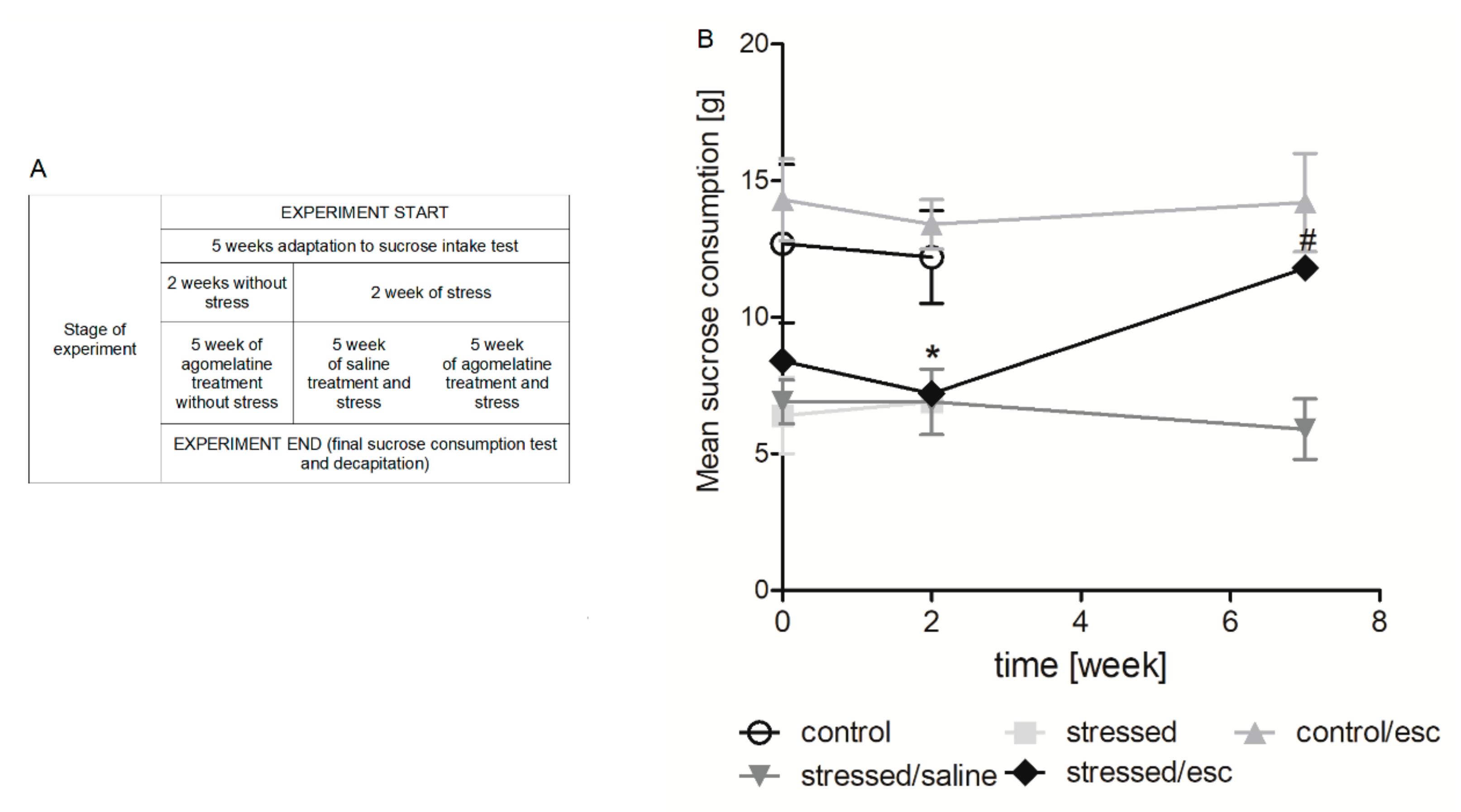
2.1. The Effect of CMS Procedure and Escitalopram Treatment on Sucrose Intake
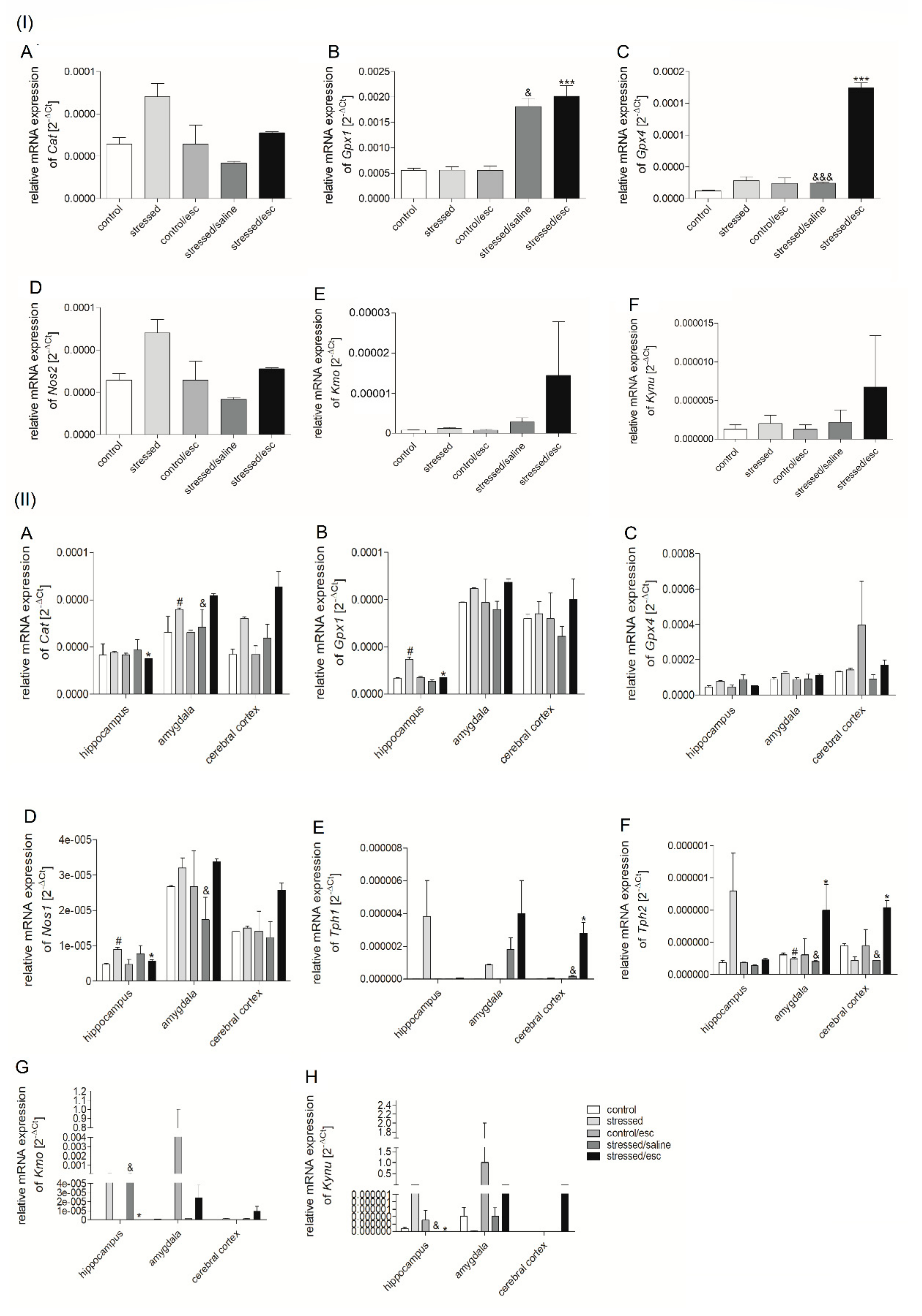
2.2. mRNA Expression
2.2.1. Gene Expression in PBMCs
2.2.2. Gene Expression in Brain Structures
2.2.3. The Effect of Escitalopram Treatment on Gene Expression in PBMCs and Brain Structures
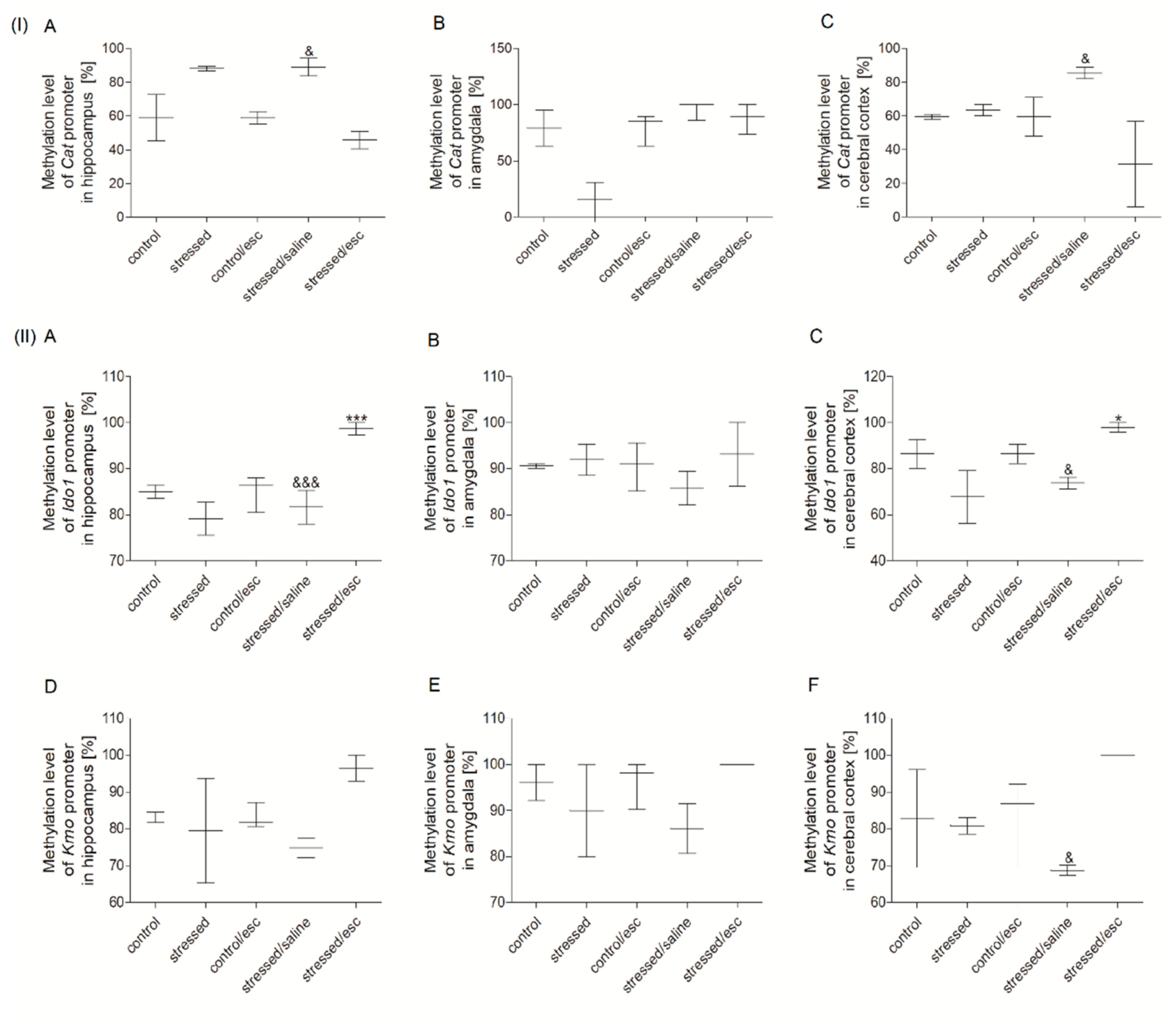
2.3. Methylation Status
2.3.1. Methylation Status of Promoter Regions in PBMCs
2.3.2. Methylation Status of Promoter Regions in Brain Structures
2.3.3. The Effect of Escitalopram Treatment on the Methylation Status of Promoter Regions in PBMCs and Brain Structures
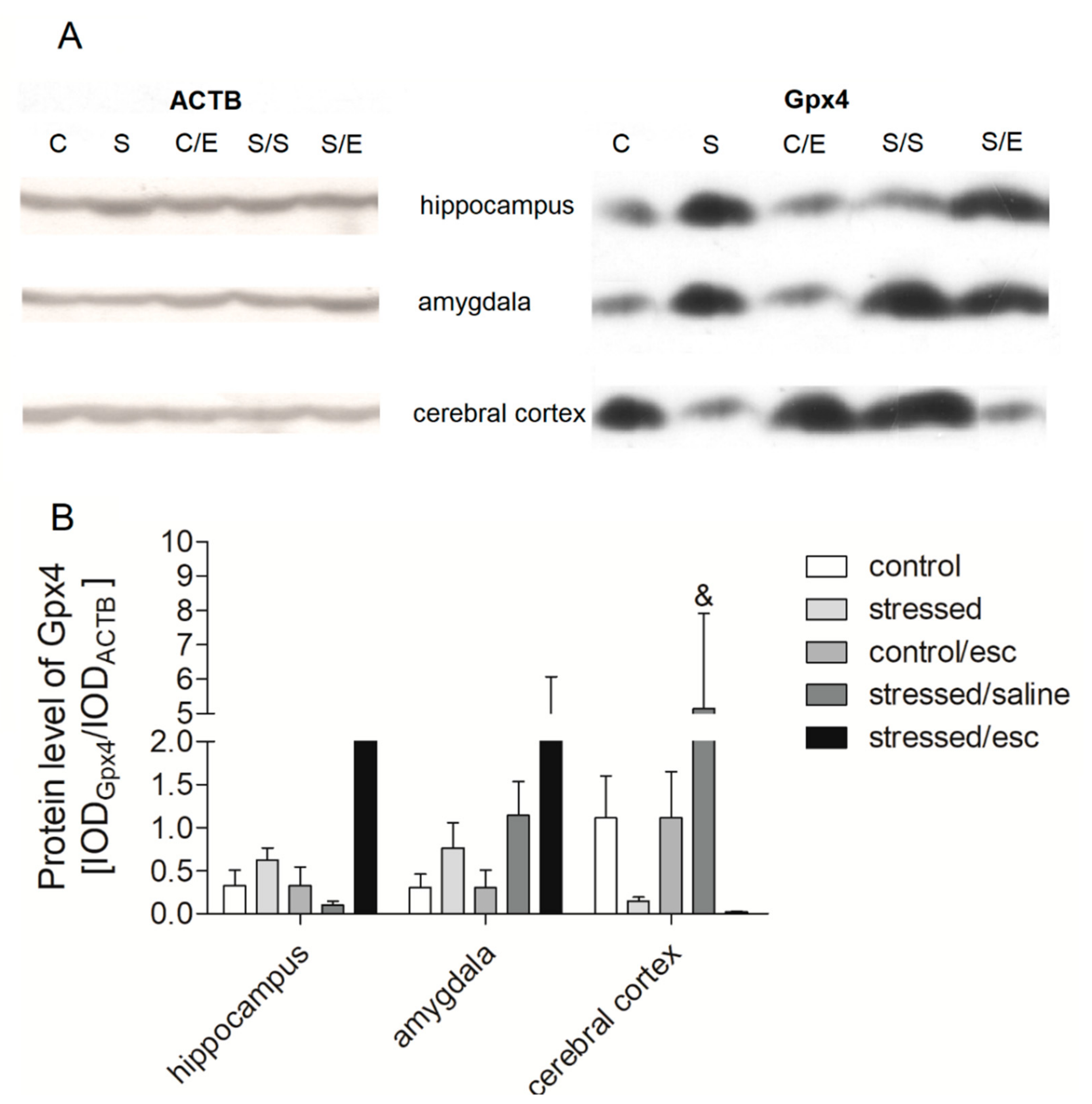
2.4. Protein Expression in Brain Structures
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. CMS
5.3. Drug
5.4. Collection of PBMCs and Brain Structure Specimens
5.5. Determination of mRNA Expression Level in PBMCs and Brain Structures
5.6. Determination of Methylation Status in PBMCs and Brain Structures
5.7. Determination of Protein Expression in the Tested Brain Structures
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar]
- Seedat, S.; Scott, K.M.; Angermeyer, M.C.; Berglund, P.; Bromet, E.J.; Brugha, T.S.; Demyttenaere, K.; De Girolamo, G.; Haro, J.M.; Jin, R.; et al. Cross-National Associations Between Gender and Mental Disorders in the World Health Organization World Mental Health Surveys. Arch. Gen. Psychiatry 2009, 66, 785–795. [Google Scholar] [CrossRef]
- Marcus, M.; Yasamy, M.T.; Van Van Ommeren, M.; Chisholm, D.; Saxena, S. Depression: A Global Public Health Concern. Social Psychiatry Psychiatr. Epidemiol. 2012, 1, 6–8. [Google Scholar]
- Depression. Available online: http://www.who.int/news-room/fact-sheets/detail/depression (accessed on 1 March 2020).
- Di Luca, M.; Olesen, J. The Cost of Brain Diseases: A Burden or a Challenge? Neuron 2014, 82, 1205–1208. [Google Scholar] [CrossRef]
- Al-Harbi, K.S. Treatment-resistant depression: Therapeutic trends, challenges, and future directions. Patient Prefer. Adherence 2012, 6, 369–388. [Google Scholar] [CrossRef]
- Wigner, P.; Czarny, P.; Galecki, P.; Su, K.-P.; Śliwiński, T. The molecular aspects of oxidative & nitrosative stress and the tryptophan catabolites pathway (TRYCATs) as potential causes of depression. Psychiatry Res. 2018, 262, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Wigner, P.; Czarny, P.; Galecki, P.; Sliwinski, T. Oxidative and nitrosative stress as well as the tryptophan catabolites pathway in depressive disorders. Psychiatr. Danub. 2017, 29, 394–400. [Google Scholar] [CrossRef]
- Dykens, J.A.; Sullivan, S.G.; Stern, A. Oxidative reactivity of the tryptophan metabolites 3-hydroxyanthranilate, cinnabarinate, quinolinate and picolinate. Biochem. Pharmacol. 1987, 36, 211–217. [Google Scholar] [CrossRef]
- Ríos, C.; Santamaría, A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem. Res. 1991, 16, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 2002, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–3863. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.E.; Leopold, M.C.; Huang, X.; Atwood, C.S.; Saunders, A.J.; Hartshorn, M.; Lim, J.T.; Faget, K.Y.; Muffat, J.A.; Scarpa, R.C.; et al. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid Generate Hydrogen Peroxide and Promote α-Crystallin Cross-Linking by Metal Ion Reduction. Biochemistry 2000, 39, 7266–7275. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Haneda, M.; Yoshino, M. Prooxidant action of xanthurenic acid and quinoline compounds: Role of transition metals in the generation of reactive oxygen species and enhanced formation of 8-hydroxy-2′-deoxyguanosine in DNA†. BioMetals 2006, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, A.; Galván-Arzate, S.; Lisý, V.; Ali, S.F.; Duhart, H.M.; Osorio-Rico, L.; Rıos, C.; Sut’Astný, F. Quinolinic acid induces oxidative stress in rat brain synaptosomes. NeuroReport 2001, 12, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Smith, R.A.; Stone, T.W. 5-Hydroxyanthranilic Acid, a Tryptophan Metabolite, Generates Oxidative Stress and Neuronal Death via p38 Activation in Cultured Cerebellar Granule Neurones. Neurotox. Res. 2009, 15, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Pandya, C.D.; Howell, K.R.; Pillai, A. Antioxidants as potential therapeutics for neuropsychiatric disorders. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2013, 46, 214–223. [Google Scholar] [CrossRef]
- Bilici, M.; Efe, H.; Köroğlu, M.; Uydu, H.A.; Bekaroğlu, M.; Değer, O. Antioxidative enzyme activities and lipid peroxidation in major depression: Alterations by antidepressant treatments. J. Affect. Disord. 2001, 64, 43–51. [Google Scholar] [CrossRef]
- Herken, H.; Gurel, A.; Selek, S.; Armutcu, F.; Ozen, M.E.; Bulut, M.; Kap, O.; Yumru, M.; Savas, H.A.; Akyol, O. Adenosine Deaminase, Nitric Oxide, Superoxide Dismutase, and Xanthine Oxidase in Patients with Major Depression: Impact of Antidepressant Treatment. Arch. Med Res. 2007, 38, 247–252. [Google Scholar] [CrossRef]
- Galecki, P.; Szemraj, J.; Bienkiewicz, M.; Florkowski, A.; Galecka, E. Lipid peroxidation andantioxidant protection in patients during acute depressive episodes and in remission after fluoxetine treatment. Pharmacol. Rep. 2009, 61, 436–447. [Google Scholar] [CrossRef]
- Kodydková, J.; Vávrová, L.; Zeman, M.; Jirák, R.; Macásek, J.; Stanková, B.; Tvrzická, E.; Zák, A. Antioxidative enzymes and increased oxidative stress in depressive women. Clin. Biochem. 2009, 42, 1368–1374. [Google Scholar] [CrossRef]
- Kotan, V.O.; Sarandol, E.; Kirhan, E.; Ozkaya, G.; Kirli, S. Effects of long-term antidepressant treatment on oxidative status in major depressive disorder: A 24-week follow-up study. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2011, 35, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Hastings, R.S.; Parsey, R.V.A.; Oquendo, M.; Arango, V.; Mann, J.J. Volumetric Analysis of the Prefrontal Cortex, Amygdala, and Hippocampus in Major Depression. Neuropsychopharmacology 2003, 29, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Jain, S. Faculty Opinions recommendation of Depression-related variation in brain morphology over 3 years: Effects of stress? Fac. Opin. Post Publ. Peer Rev. Biomed. Lit. 2008, 65, 1156–1165. [Google Scholar] [CrossRef]
- Campbell, S.; MacQueen, G. The role of the hippocampus in the pathophysiology of major depression. J. Psychiatry Neurosci. 2004, 29, 417–426. [Google Scholar]
- Pulschen, D.; Thome, J. The Role of Oxidative Stress in Depressive Disorders. Curr. Pharm. Des. 2012, 18, 5890–5899. [Google Scholar] [CrossRef]
- Shalaby, A.; Kamal, S. Effect of Escitalopram on GABA level and anti-oxidant markers in prefrontal cortex and nucleus accumbens of chronic mild stress-exposed albino rats. Int. J. Physiol. Pathophysiol. Pharmacol. 2009, 1, 154–161. [Google Scholar]
- Sarandol, A.; Sarandol, E.; Eker, S.S.; Erdinc, S.; Vatansever, E.; Kirli, S. Major depressive disorder is accompanied with oxidative stress: Short-term antidepressant treatment does not alter oxidative–antioxidative systems. Hum. Psychopharmacol. Clin. Exp. 2007, 22, 67–73. [Google Scholar] [CrossRef]
- Cimen, A.P.B.; Gumus, C.B.; Cetin, A.P.I.; Ozsoy, A.P.S.; Aydin, M.; Cimen, L. The Effects of Escitalopram Treatment on Oxidative/ Antioxidative Parameters in Patients with Depression. Klinik Psikofarmakol. Bülteni Bull. Clin. Psychopharmacol. 2015, 25, 272–279. [Google Scholar] [CrossRef]
- Duda, W.; Curzytek, K.; Kubera, M.; Iciek, M.; Kowalczyk-Pachel, D.; Bilska-Wilkosz, A.; Lorenc-Koci, E.; Leskiewicz, M.; Basta-Kaim, A.; Budziszewska, B.; et al. The Effect of Chronic Mild Stress and Imipramine on the Markers of Oxidative Stress and Antioxidant System in Rat Liver. Neurotox. Res. 2016, 30, 173–184. [Google Scholar] [CrossRef]
- Liu, T.; Zhong, S.; Liao, X.; Chen, J.; He, T.; Lai, S.; Jia, Y. A Meta-Analysis of Oxidative Stress Markers in Depression. PLoS ONE 2015, 10, e0138904. [Google Scholar] [CrossRef]
- Ahmadimanesh, M.; Abbaszadegan, M.R.; Rad, D.M.; Moallem, S.M.H.; Mohammadpour, A.H.; Ghahremani, M.H.; Hosseini, F.F.; Behdani, F.; Manteghi, A.A.; Jowsey, P.; et al. Effects of selective serotonin reuptake inhibitors on DNA damage in patients with depression. J. Psychopharmacol. 2019, 33, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Tohid, H.; Aleem, D.; Jackson, C. Major Depression and Psoriasis: A Psychodermatological Phenomenon. Ski. Pharmacol. Physiol. 2016, 29, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Gamaro, G.D.; Streck, E.L.; Matté, C.; Prediger, M.E.; Wyse, A.T.S.; Dalmaz, C. Reduction of hippocampal Na+, K+-ATPase activity in rats subjected to an experimental model of depression. Neurochem. Res. 2003, 28, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Bekris, S.; Antoniou, K.; Daskas, S.; Papadopoulou-Daifoti, Z. Behavioural and neurochemical effects induced by chronic mild stress applied to two different rat strains. Behav. Brain Res. 2005, 161, 45–59. [Google Scholar] [CrossRef]
- Papp, M. Models of Affective Illness: Chronic Mild Stress in the Rat. Curr. Protoc. Pharmacol. 2012, 57, 5–9. [Google Scholar] [CrossRef]
- Papp, M.; Gruca, P.; Lason-Tyburkiewicz, M.; Litwa, E.; Niemczyk, M.; Tota-Glowczyk, K.; Willner, P. Dopaminergic mechanisms in memory consolidation and antidepressant reversal of a chronic mild stress-induced cognitive impairment‘. Psychopharmacology 2017, 234, 2571–2585. [Google Scholar] [CrossRef]
- Papp, M.; Gruca, P.; Lason, M.; Niemczyk, M.; Willner, P. The role of prefrontal cortex dopamine D2 and D3 receptors in the mechanism of action of venlafaxine and deep brain stimulation in animal models of treatment-responsive and treatment-resistant depression. J. Psychopharmacol. 2019, 33, 748–756. [Google Scholar] [CrossRef]
- Wang, Q.; Timberlake, M.A., 2nd; Prall, K.; Dwivedi, Y. The recent progress in animal models of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 99–109. [Google Scholar] [CrossRef]
- Tanke, M.A.C.; Alserda, E.; Doornbos, B.; Van Der Most, P.J.; Goeman, K.; Postema, F.; Korf, J. Low tryptophan diet increases stress-sensitivity, but does not affect habituation in rats. Neurochem. Int. 2008, 52, 272–281. [Google Scholar] [CrossRef]
- Jacobsen, J.P.R.; Siesser, W.B.; Sachs, B.D.; Peterson, S.; Cools, M.J.; Setola, V.; Folgering, J.H.A.; Flik, G.; Caron, M.G. Deficient serotonin neurotransmission and depression-like serotonin biomarker alterations in tryptophan hydroxylase 2 (Tph2) loss-of-function mice. Mol. Psychiatry 2011, 17, 694–704. [Google Scholar] [CrossRef]
- Wigner, P.; Synowiec, E.; Czarny, P.; Bijak, M.; Jóźwiak, P.; Szemraj, J.; Gruca, P.; Papp, M.; Śliwiński, T. Effects of venlafaxine on the expression level and methylation status of genes involved in oxidative stress in rats exposed to a chronic mild stress. J. Cell. Mol. Med. 2020, 24, 5675–5694. [Google Scholar] [CrossRef] [PubMed]
- Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Bijak, M.; Białek, K.; Szemraj, J.; Gruca, P.; Papp, M.; Śliwiński, T. The Effect of Chronic Mild Stress and Venlafaxine on the Expression and Methylation Levels of Genes Involved in the Tryptophan Catabolites Pathway in the Blood and Brain Structures of Rats. J. Mol. Neurosci. 2020, 70, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Moylan, S.; Berk, M.; Dean, O.M.; Samuni, Y.; Williams, L.J.; O’Neil, A.; Hayley, A.C.; Pasco, J.A.; Anderson, G.M.; Jacka, F.N.; et al. Oxidative & nitrosative stress in depression: Why so much stress? Neurosci. Biobehav. Rev. 2014, 45, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.-M. Kynurenines: From the perspective of major psychiatric disorders. FEBS J. 2012, 279, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef]
- Lucca, G.; Comim, C.M.; Valvassori, S.S.; Réus, G.Z.; Vuolo, F.; Petronilho, F.; Gavioli, E.C.; Dal-Pizzol, F.; Quevedo, J. Increased oxidative stress in submitochondrial particles into the brain of rats submitted to the chronic mild stress paradigm. J. Psychiatr. Res. 2009, 43, 864–869. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Shigenaga, M.K.; Yeo, H.C.; Mori, A.; Ames, B.N. Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J. 1996, 10, 1532–1538. [Google Scholar] [CrossRef]
- Qiu, H.-M.; Yang, J.-X.; Jiang, X.-H.; Hu, X.-Y.; Liu, D.; Zhou, Q.-X. Enhancing tyrosine hydroxylase and tryptophan hydroxylase expression and improving oxidative stress involved in the antidepressant effect of sodium valproate on rats undergoing chronic unpredicted stress. NeuroReport 2015, 26, 1145–1150. [Google Scholar] [CrossRef]
- Muller, C.P.; Jacobs, B.L. Handbook of the Behavioral Neurobiology of Serotonin; Academic Press: London, UK, 2010. [Google Scholar]
- Nakamura, K.; Hasegawa, H. Developmental role of tryptophan hydroxylase in the nervous system. Mol. Neurobiol. 2007, 35, 45–53. [Google Scholar] [CrossRef]
- Cowen, P.J.; Browning, M. What has serotonin to do with depression? World Psychiatry 2015, 14, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.S.; Iradukunda, E.C.; Hughes, T.; Bowen, J.P. Modulation of Enzyme Activity in the Kynurenine Pathway by Kynurenine Monooxygenase Inhibition. Front. Mol. Biosci. 2019, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O’Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Emetz, R.; Muller, A.J.; Merlo, L.M.F.; Emandik-Nayak, L. IDO2 in Immunomodulation and Autoimmune Disease. Front. Immunol. 2014, 5, 585. [Google Scholar] [CrossRef]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–8110. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Borgbo, T.; Hansen, L.L. Primer design versus PCR bias in methylation independent PCR amplifications. Epigenetics 2009, 4, 231–234. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidative and Nitrosative Stresses | ||||
|---|---|---|---|---|
| Gene | Enzyme | Gene Location | Function of the Enzyme | Tissue mRNA Expression |
| Gpx1 | Glutathione peroxidase 1 | 8q32 | Enzyme catalyses the reduction of organic hydroperoxides and hydrogen peroxide by glutathione, and thereby protect cells against oxidative damage. | Detected in all tissue |
| Gpx4 | Glutathione peroxidase 4 | 7q11 | Enzyme which catalyses the reduction of hydrogen peroxide, organic hydroperoxides and lipid hydroperoxides, and thereby protect cells against oxidative damage. Essential antioxidant peroxidase that directly reduces phospholipid hydroperoxide even if they are incorporated in membranes and lipoproteins (By similarity). Can also reduce fatty acid hydroperoxide, cholesterol hydroperoxide and thymine hydroperoxide. | Detected in all tissue |
| Cat | Catalase | 3q32 | The key antioxidant enzyme in the bodies defence against oxidative stress. Catalase is a heme enzyme that is present in the peroxisome of nearly all aerobic cells. Catalase converts the reactive oxygen species hydrogen peroxide to water and oxygen and thereby mitigates the toxic effects of hydrogen peroxide. | Detected in all tissue, however tissue enhanced–blood and liver |
| Nos1 | Nitric oxide synthetase 1 | 12q16 | Enzyme, which synthesize nitric oxide from L-arginine. | Brain, skeletal muscle |
| Nos2 | Nitric oxide synthetase 2 | 10q25 | Enzyme, which synthesize nitric oxide from L-arginine. | Detected in many tissue, however tissue enhanced–intestine, lymphoid tissue |
| Tryptophan catabolites pathway | ||||
| Gene | Enzyme | Gene location | Function of the enzyme | Tissue mRNA expression |
| Tph1 | Tryptophan hydroxylase 1 | 1q22 | The enzyme catalyses the first and rate limiting step in the biosynthesis of serotonin, an important hormone and neurotransmitter. | Brain, intestine, pituitary gland, stomach |
| Tph2 | Tryptophan hydroxylase 2 | 7q22 | The encoded protein catalyses the first and rate limiting step in the biosynthesis of serotonin, an important hormone and neurotransmitter. | Brain |
| Ido1 | Indolamine 2,3-dioxygenasse | 16q12.5 | Ido1 is heme enzyme that catalyses the first and rate-limiting step in tryptophan catabolism to N-formyl-kynurenine. This enzyme acts on multiple tryptophan substrates including D-tryptophan, L-tryptophan, 5-hydroxy-tryptophan, tryptamine, and serotonin. | Blood, placenta |
| Kmo | Kynurenine 3-monooxygenase | 13q25 | The enzyme catalyses the hydroxylation of L-kynurenine to form 3-hydroxy-L-kynurenine. It is Required for synthesis of quinolinic acid, a neurotoxic NMDA receptor antagonist and potential endogenous inhibitor of NMDA receptor signalling. | Blood, liver, placenta |
| Kynu | Kynureninase | 3q12 | It is enzyme that catalyses the cleavage of L-kynurenine and L-3-hydroxykynurenine into anthranilic and 3-hydroxyanthranilic acids, respectively. Kynureninase is involved in the biosynthesis of NAD cofactors from tryptophan through the kynurenine pathway | Detected in all tissue, however tissue enhanced–blood, liver, placenta |
| Oxidative and Nitrosative Stresses | |||||||||||
| Gene | Starter Sequence (5′->3′) | Product Size [bp] | The Condition of the Reaction | ||||||||
| The Initial Activation of the Polymerase | Denaturation | Annealing | Elongation | The HRM Analysis | |||||||
| Temperature [°C] | Time [min] | Temperature [°C] | Time [s] | Temperature [°C] | Time [s] | Temperature [°C] | Time [s] | ||||
| Cat | F: TTTGAGATTATTGTGTTTGAAA R: TACCTACACCCAAAAAAAAATA | 148 | 95 | 12 | 95 | 15 | 59 | 20 | 72 | 20 | Denaturation at 95 °C for 15 s, reannealing at 60 °C for 1 min and melting from 60 to 95 °C at a ramp rate of 0.2 °C |
| Gpx1 | F: GTTGTTTTAGGTTTTGTTGTTG R: AAAACTAAAATCCTCCAACTCT | 102 | 65 | ||||||||
| Gpx4 (promotor 2) | F: AGGTTGGAGGTTTAGAGGTTTA R: TCCCCTAAATACAAAAATCTCT | 118 | 59 | ||||||||
| Gpx4 (promotor 3) | F: AGGTTGGAGGTTTAGAGGTTTA R: AAAACATAACAAAATCATCTCCC | 147 | 65 | ||||||||
| Nos1 (promotor 5) | F: GGGTTTTTAATTTTTTTATTGTG R: CAACCCTCATTAAAAAAACC | 124 | 59 | ||||||||
| Nos1 (promotor 7) | F: GTTTGAGATTGGAATTTTTTGG R: CCAAAACATCCAAAAATACACA | 124 | 59 | ||||||||
| Tryptophan catabolites pathway | |||||||||||
| Gene | Starter sequence (5′->3′) | Product size [bp] | The condition of the reaction | ||||||||
| The initial activation of the polymerase | Denaturation | Annealing | Elongation | The HRM analysis | |||||||
| Temperature [°C] | Time [min] | Temperature [°C] | Time [s] | Temperature [°C] | Time [s] | Temperature [°C] | Time [s] | ||||
| Tph1 | F: GGGAGTTTTGTTTTGGTTTTTA R: TCCTCAACCACAAAAAATCTAA | 132 | 95 | 12 | 95 | 15 | 55 | 20 | 72 | 20 | Denaturation at 95 °C for 15 s, reannealing at 60 °C for 1 min and melting from 60 to 95 °C at a ramp rate of 0.2 °C |
| Ido1 | F: TTTGAGTTTTAGTGATTTTGGG R: TTAATATCTAATCCCAATCTCTAAAAC | 100 | 59 | ||||||||
| Tdo2 (promoter 1) | F: GATGATTTAGGTGGTTTGAGGT R: CAAAAAAAACAAAATTCATCCA | 123 | 59 | ||||||||
| Tdo2 (promoter 2) | F: ATGATTTAGGTGGTTTGAGGTT R: ACCCAATCTACCTAACTAACAAC | 187 | 61.4 | ||||||||
| Kmo | F: TTGGTTTAGGGAAGGAAATR: ATAAAAAACTAAACCCAAAACAC | 150 | 55.7 | ||||||||
| Oxidative Stress | ||||||||
| Protein | Primary Antibody | Secondary Antibody | ||||||
| Producent | The Origin of Antibodies | Dilution | Condition of Incubation [h] | Producent | The Origin of Antibodies | Dilution | Condition of Incubation [h] | |
| β-actin (a reference protein) | Santa Cruz Biotechnology Inc, Dallas, Texas, USA | Mouse | 1:1000 | 1 h at room temperature | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-mouse | 1:6000 | 1 h at room temperature |
| catalase | Santa Cruz Biotechnology Inc, Dallas, Texas, USA | Mouse | 1:1000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-mouse | 1:6000 | 1 h at room temperature |
| glutathione peroxidase 4 | Abcam, Cambridge, United Kingdom, | Rabbit | 1:6000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-rabbit | 1:6000 | 1 h at room temperature |
| superoxide dismutase 1 | Santa Cruz Biotechnology Inc, Dallas, Texas, USA | Mouse | 1:6000 | 2 h at room temperature | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-mouse | 1:6000 | 1 h at room temperature |
| Tryptophan catabolites pathway | ||||||||
| Protein | Primary antibody | Secondary antibody | ||||||
| Producent | The origin of antibodies | Dilution | Condition of incubation [h] | Producent | The origin of antibodies | Dilution | Condition of incubation [h] | |
| Tryptophan hydroxylase 1 | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Rabbit | 1:1000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-rabbit | 1:6000 | 1 h at room temperature |
| Tryptophan hydroxylase 2 | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Rabbit | 1:6000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-rabbit | 1:6000 | 1 h at room temperature |
| Indoleamine 2,3-dioxygenase | Santa Cruz Biotechnology Inc, Dallas, Texas, USA | Mouse | 1:1000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-rabbit | 1:6000 | 1 h at room temperature |
| Kynurenine aminotransferases | Santa Cruz Biotechnology Inc, Dallas, Texas, USA | Mouse | 1:1000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-rabbit | 1:6000 | 1 h at room temperature |
| Kynureninase | Santa Cruz Biotechnology Inc, Dallas, Texas, USA | Mouse | 1:1000 | overnight at 4 °C | Cell Signalling Technologies Inc., Danvers, Massachusetts, USA | Anti-rabbit | 1:6000 | 1 h at room temperature |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Bijak, M.; Białek, K.; Szemraj, J.; Gruca, P.; Papp, M.; Śliwiński, T. The Effect of Chronic Mild Stress and Escitalopram on the Expression and Methylation Levels of Genes Involved in the Oxidative and Nitrosative Stresses as Well as Tryptophan Catabolites Pathway in the Blood and Brain Structures. Int. J. Mol. Sci. 2021, 22, 10. https://doi.org/10.3390/ijms22010010
Wigner P, Synowiec E, Jóźwiak P, Czarny P, Bijak M, Białek K, Szemraj J, Gruca P, Papp M, Śliwiński T. The Effect of Chronic Mild Stress and Escitalopram on the Expression and Methylation Levels of Genes Involved in the Oxidative and Nitrosative Stresses as Well as Tryptophan Catabolites Pathway in the Blood and Brain Structures. International Journal of Molecular Sciences. 2021; 22(1):10. https://doi.org/10.3390/ijms22010010
Chicago/Turabian StyleWigner, Paulina, Ewelina Synowiec, Paweł Jóźwiak, Piotr Czarny, Michał Bijak, Katarzyna Białek, Janusz Szemraj, Piotr Gruca, Mariusz Papp, and Tomasz Śliwiński. 2021. "The Effect of Chronic Mild Stress and Escitalopram on the Expression and Methylation Levels of Genes Involved in the Oxidative and Nitrosative Stresses as Well as Tryptophan Catabolites Pathway in the Blood and Brain Structures" International Journal of Molecular Sciences 22, no. 1: 10. https://doi.org/10.3390/ijms22010010
APA StyleWigner, P., Synowiec, E., Jóźwiak, P., Czarny, P., Bijak, M., Białek, K., Szemraj, J., Gruca, P., Papp, M., & Śliwiński, T. (2021). The Effect of Chronic Mild Stress and Escitalopram on the Expression and Methylation Levels of Genes Involved in the Oxidative and Nitrosative Stresses as Well as Tryptophan Catabolites Pathway in the Blood and Brain Structures. International Journal of Molecular Sciences, 22(1), 10. https://doi.org/10.3390/ijms22010010