Cytokine Release Syndrome in COVID-19 Patients, A New Scenario for an Old Concern: The Fragile Balance between Infections and Autoimmunity

 , , and
, , and 
Abstract
1. Introduction
2. SARS-CoV-2 Infection and the Pathophysiology of Cytokine Release Syndrome
3. Anti-Host Therapy in SARS-CoV-2 Patients
4. Chloroquine and Hydroxychloroquine
5. IL-6 Inhibitors
6. IL1-Inhibitors
7. JAK-STAT Inhibitors
8. TNF-α Inhibitors
9. Other Potential Immunological Therapeutic Options
9.1. B Cell Inhibitors
9.2. T Cell Modulation Therapy
10. Autoimmunity and SARS-CoV-2 Infection
11. Conclusions
Authors Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in china, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef] [PubMed]
- John Hopkins University of Medicine; coronavirus resource center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 7 April 2020).
- Jiang, F.; Deng, L.; Zhang, L.; Cai, Y.; Cheung, C.-W.; Xia, Z. Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J. Gen. Intern. Med. 2020, 1–5. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; Von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Chatenoud, L.; Ferran, C.; Reuter, A.; Legendre, C.; Gevaert, Y.; Kreis, H.; Franchimont, P.; Bach, J.F. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N. Engl. J. Med. 1989, 320, 1420–1421. [Google Scholar]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Murthy, H.S.; Iqbal, M.; Chavez, J.C.; Kharfan-Dabaja, M.A. Cytokine release syndrome: Current perspectives. ImmunoTargets Ther. 2019, 8, 43–52. [Google Scholar] [CrossRef]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.B.; Hoang, D.M.; Chau, N.V.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Rosário, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.; Shoenfeld, Y. The Hyperferritinemic Syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Invernizzi, P.; Arosio, P.; Cairo, G. New functions for an iron storage protein: The role of ferritin in immunity and autoimmunity. J. Autoimmun. 2008, 30, 84–89. [Google Scholar] [CrossRef]
- Betancur, J.-F.; Navarro, E.-P.; Echeverry, A.; Moncada, P.A.; Cañas, C.A.; Tobón, G.J. Hyperferritinemic syndrome: Still’s disease and catastrophic antiphospholipid syndrome triggered by fulminant Chikungunya infection: A case report of two patients. Clin. Rheumatol. 2015, 34, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.-L.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef]
- Demirkol, D.; Yıldızdaş, D.; Bayrakcı, B.; Karapinar, B.; Kendirli, T.; Koroglu, T.F.; Dursun, O.; Erkek, N.; Gedik, A.H.; Çıtak, A.; et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: What is the treatment? Crit. Care 2012, 16, R52. [Google Scholar] [CrossRef]
- Kernan, K.F.; Joseph, A. Carcillo. Hyperferritinemia and inflammation. Int. Immunol. 2017, 29, 401–409. [Google Scholar] [CrossRef]
- Gray, C.P.; Arosio, P.; Hersey, P. Telford, R. Heavy chain ferritin activates regulatory T cells by induction of changes in dendritic cells. Blood 2002, 99, 3326–3334. [Google Scholar] [CrossRef]
- Carcillo, J.A.; Sward, K.; Halstead, E.S.; Jimenez-Bacardi, A.; Shakoory, B.; Simon, D.; Hall, M. A systemic inflammation mortality risk assessment contingency table for severe sepsis. Pediatr. Crit. Care Med. 2017, 18, 143. [Google Scholar] [CrossRef]
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893–2897. [Google Scholar] [CrossRef]
- Dinkla, S.; van Eijk, L.T.; Fuchs, B.; Schiller, J.; Joosten, I.; Brock, R.; Pickkers, P.; Bosman, G.J. Inflammation associated changes in lipid composition and the organization of the erythrocyte membrane. BBA Clin. 2016, 5, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.; Eubanks, S.K.; Schaffer, K.J.; Zhou, C.Y.; Linder, M.C. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood 1997, 90, 4979–4986. [Google Scholar] [CrossRef] [PubMed]
- Colafrancesco, S.; Priori, R.; Alessandri, C.; Astorri, E.; Perricone, C.; Blank, M.; Agmon-Levin, N.; Shoenfeld, Y.; Valesini, G. The Hyperferritinemic Syndromes and CD163: A Marker of Macrophage Activation. IMAJ 2014, 16, 662–663. [Google Scholar] [PubMed]
- Bleesing, J.; Prada, A.; Siegel, D.M.; Villanueva, J.; Olson, J.; Ilowite, N.T.; Brunner, H.I.; Griffin, T.; Graham, T.B.; Sherry, D.D.; et al. The Diagnostic Significance of Soluble CD163 and Soluble Interleukin-2 Receptor -Chain in Macrophage Activation Syndrome and Untreated New-Onset Systemic Juvenile Idiopathic Arthritis. Arthritis Rheum. 2007, 56, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lam, C.W.K.; Wu, A.K.L.; Ip, W.K.; Lee, N.; Chan, I.H.S.; Lit, L.C.W.; Hui, D.S.; Chan, M.H.M.; Chung, S.S.C.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- Romagnani, S. Regulation of the T cell response. Clin. Exp. Allergy 2006, 36, 1357–1366. [Google Scholar] [CrossRef]
- Cilloniz, C.; Pantin-Jackwood, M.; Ni, C.; Goodman, A.G.; Peng, X.; Proll, S.C.; Carter, V.S.; Rosenzweig, E.R.; Szretter, K.; Katz, J.M.; et al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J. Virol. 2010, 84, 7613–7624. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Liu, H.M.; Chang, M.-F.; Chang, S.C. Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses type I and type III interferon induction by targeting RIG-I signaling. J. Virol. 2020. [Google Scholar] [CrossRef]
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. Ifn-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J. Exp. Med. 2015, 212, 845–853. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Alphonse, N.; Dickenson, R.E.; Durbin, J.E.; Glenn, J.S.; Hartmann, R.; Kotenko, S.V.; LaZear, H.M.; O’Brien, T.R.; Odendall, C.; et al. COVID-19 and emerging viral infections: The case for interferon lambda. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Frazier, W.J.; Hall, M.W. Immunoparalysis and adverse outcomes from critical illness. Pediatr. Clin. N. Am. 2008, 55, 647–668. [Google Scholar] [CrossRef] [PubMed]
- Monneret, G.; Lepape, A.; Voirin, N.; Bohé, J.; Venet, F.; Debard, A.-L.; Thizy, H.; Bienvenu, J.; Gueyffier, F.; Vanhems, P. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensiv. Care Med. 2006, 32, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Chen, L.; Li, J.; Wang, X.; Wang, F.; et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv 2020. [Google Scholar] [CrossRef]
- Obstfeld, A.E.; Frey, N.V.; Mansfield, K.; Lacey, S.F.; June, C.H.; Porter, D.L.; Melenhorst, J.J.; Wasik, M.A. Cytokine release syndrome associated with chimeric-antigen receptor T-cell therapy: Clinicopathological insights. Blood 2017, 130, 2569–2572. [Google Scholar] [CrossRef]
- Cossarizza, A.; De Biasi, S.; Guaraldi, G.; Girardis, M.; Mussini, C.; The Modena Covid-19 Working Group (MoCo19). SARS-CoV-2, the virus that causes COVID-19: Cytometry and the new challenge for global health. Cytom. Part A 2020, 97, 340–343. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Ye, L.; Chen, X.; Li, R.; Pan, Z.; Qian, C.; Yang, Y.; You, R.; Zhao, J.; Gao, L.; Li, Z.; et al. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Lan, L.; Xu, D.; Ye, G.; Xia, C.; Wang, S.; Li, Y.; Xu, H. Positive RT-PCR test results in patients recovered from COVID-19. JAMA 2020. [Google Scholar] [CrossRef]
- Ng, O.-W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hou, J.; Jiang, X.; Ma, S.; Meng, M.; Wang, B.; Zhang, M.; Zhang, M.; Tang, X.; Zhang, F.; et al. Response of memory CD8+ T cells to severe acute respiratory syndrome (SARS) coronavirus in recovered SARS patients and healthy individuals. J. Immunol. 2005, 175, 591–598. [Google Scholar] [CrossRef]
- Tang, F.; Quan, Y.; Xin, Z.; Wrammert, J.; Ma, M.-J.; Lv, H.; Wang, T.-B.; Yang, H.; Richardus, J.H.; Liu, W.; et al. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: A six-year follow-up study. J. Immunol. 2011, 186, 7264–7268. [Google Scholar] [CrossRef]
- Clinical Management of Severe Acute Respiratory Infection When COVID-19 Is Suspected. Available online: https://www.who.int/publications-detail/clinical-management-of-severe-acute-respiratory-infection-when-novel-coronavirus-(ncov)-infection-is-suspected (accessed on 7 May 2020).
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the alert for cytokine storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID-19) infected patients. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.; Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Lee, B.L.; Lau, L.; Wickliffe, K.E.; Shi, G.-P.; Chapman, H.A.; Barton, G.M. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 2008, 456, 658–662. [Google Scholar] [CrossRef]
- Van der Borne, B.E.; Dijkmans, B.A.; De Rooij, H.H.; Le Cessie, S.; Verweij, C.L. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J. Rheumatol. 1997, 24, 55–60. [Google Scholar]
- Belizna, C.; Pregnolato, F.; Abad, S.; Alijotas-Reig, J.; Amital, H.; Amoura, Z.; Andreoli, L.; Andrès, E.; Aouba, A.; Bilgen, S.A.; et al. HIBISCUS: Hydroxychloroquine for the secondary prevention of thrombotic and obstetrical events in primary antiphospholipid syndrome. Autoimmun. Rev. 2018, 17, 1153–1168. [Google Scholar] [CrossRef]
- Basta, F.; Irace, R.; Borgia, A.; Messiniti, V.; Riccardi, A.; Valentini, G.; Afeltra, A. Hydroxychloroquine significantly reduces serum markers of endothelial injury and NEMO videocapillaroscopy score in systemic sclerosis. Rheumatology 2019, 58, 1303–1305. [Google Scholar] [CrossRef]
- Keyaerts, E.; Vijgen, L.; Maes, P.; Neyts, J.; Van Ranst, M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 2004, 323, 264–268. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, É.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020. [Google Scholar] [CrossRef]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef]
- Linee Guida Sulla Gestione Terapeutica e di Supporto per Pazienti con Infezione da Coronavirus COVID-19—Società Italiana di Malattie Infettive e Tropicali. Available online: http://www.fvcalabria.unicz.it/COVID-19/LINEE-GUIDA/linee-guida-SIMIT-marzo-2020.pdf (accessed on 2 April 2020).
- Tanaka, T.; Kishimoto, T. Targeting Interleukin-6: All the way to treat autoimmune and inflammatory diseases. Int. J. Biol. Sci. 2012, 8, 1227–1236. [Google Scholar] [CrossRef]
- Choy, E.; Rose-John, S. Interleukin-6 as a multifunctional regulator: inflammation, immune response, and fibrosis. J. Scleroderma Relat. Disord. 2017, 2, S1–S5. [Google Scholar] [CrossRef]
- Diamanti, A.P.; Markovic, M.; Argento, G.; Giovagnoli, S.; Ricci, A.; Laganà, B.; D’Amelio, R. Therapeutic management of patients with rheumatoid arthritis and associated interstitial lung disease: Case report and literature review. Ther. Adv. Respir. Dis. 2016, 11, 64–72. [Google Scholar] [CrossRef]
- Manfredi, A.; Cassone, G.; Furini, F.; Gremese, E.; Venerito, V.; Atzeni, F.; Arrigoni, E.; Della Casa, G.; Cerri, S.; Govoni, M.; et al. Tocilizumab therapy in rheumatoid arthritis with interstitial lung disease: A multicenter retrospective study. Intern. Med. J. 2019. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). medRxiv 2020. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef]
- ACTEMRA (Tocilizumab) Injection–FDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125276s114lbl.pdf (accessed on 6 May 2020).
- RoActemra, INN-Tocilizumab—European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/smop/chmp-post-authorisation-summary-positive-opinion-roactemra_en-3.pdf (accessed on 6 May 2020).
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. ChinaXiv 2020, 202003, v1. [Google Scholar]
- Vademecum per la Cura Delle Persone con Malattia da COVI-19 Edizione 2.0, 13 Marzo 2020—Società Italiana di Malattie Infettive e Tropicali. Available online: http://www.simit.org/medias/1568-covid19-vademecum-20-13-marzo-2020.pdf (accessed on 6 April 2020).
- Inpatient Guidance for Treatment of Covid-19 in Adults and Children—University of Michigan. Available online: http://www.med.umich.edu/asp/pdf/adult_guidelines/COVID-19-treatment.pdf (accessed on 10 April 2020).
- Behrens, E.; Koretzky, G.A. Review: Cytokine storm syndrome: Looking toward the precision medicine era. Arthritis Rheumatol. 2017, 69, 1135–1143. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Pugin, J.; Ricou, B.; Steinberg, K.P.; Suter, P.M.; Martin, T.R. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am. J. Respir. Crit. Care Med. 1996, 153, 1850–1856. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: Reanalysis of a prior phase III trial. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Grom, A.A.; Ilowite, N.; Martini, A.; Leon, K.; Lheritier, K.; Abrams, K.; Pascual, V.; Brunner, H.I.; Lovell, D.; Ruperto, N.; et al. Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with canakinumab. Arthritis Rheumatol. 2015, 68, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Kontzias, A.; Kotlyar, A.; Laurence, A.; Changelian, P.; O’Shea, J.J. Jakinibs: A new class of kinase inhibitors in cancer and autoimmune disease. Curr. Opin. Pharmacol. 2012, 12, 464–470. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.; Byers, N.L.; Higgs, R.E.; Lee, J.; Macias, W.L.; Na, S.; Ortmann, R.A.; Rocha, G.; Rooney, T.P.; Wehrman, T.; et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res. 2019, 21, 183. [Google Scholar] [CrossRef]
- Das, R.; Guan, P.; Sprague, L.; Verbist, K.C.; Tedrick, P.; An, Q.A.; Cheng, C.; Kurachi, M.; Levine, R.; Wherry, E.J.; et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood 2016, 127, 1666–1675. [Google Scholar] [CrossRef]
- Maschalidi, S.; Sepulveda, F.E.; Garrigue, A.; Fischer, A.; de Saint Basile, G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood 2016, 128, 60–71. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in inflammatory and autoimmune diseases. BioDrugs 2019, 33, 15–32. [Google Scholar] [CrossRef]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.H.; Tartaglia, L.A.; Lee, M.S.; Goeddel, D.V. Antiviral activity of tumor necrosis factor is signaled through the 55-kDa type I TNF receptor [corrected]. J. Immunol. 1992, 149, 3350–3353. [Google Scholar] [PubMed]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Maeshima, K.; Ishii, K.; Iwakura, M.; Akamine, M.; Hamasaki, H.; Abe, I.; Haranaka, M.; Tatsukawa, H.; Yoshimatsu, H. Adult-onset Still’s disease with macrophage activation syndrome successfully treated with a combination of methotrexate and etanercept. Mod. Rheumatol. 2012, 22, 137–141. [Google Scholar] [CrossRef]
- Stern, A.; Riley, R.; Buckley, L. Worsening of macrophage activation syndrome in a patient with adult onset Still’s disease after initiation of etanercept therapy. J. Clin. Rheumatol. 2001, 7, 252–256. [Google Scholar] [CrossRef]
- Keystone, E.; Fleischmann, R.; Emery, P.; Furst, D.E.; van Vollenhoven, R.; Bathon, J.; Dougados, M.; Baldassare, A.; Ferraccioli, G.; Chubick, A.; et al. Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: An open-label extension analysis. Arthritis Rheum. 2007, 56, 3896–3908. [Google Scholar] [CrossRef]
- Avilés, A.; Nambo, M.J.; Neri, N.; Cleto, S.; Castañeda, C.; Huerta-Guzmàn, J.; Murillo, E.; Contreras, M.; Talavera, A.; González, M. Dose dense (CEOP-14) vs dose dense and rituximab (CEOP-14+R) in high-risk diffuse large cell lymphoma. Med. Oncol. 2007, 24, 85–89. [Google Scholar] [CrossRef]
- MabThera, INN-rituximab—European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf (accessed on 5 May 2020).
- Weaver, L.; Behrens, E. Weathering the storm: Improving therapeutic interventions for cytokine storm syndromes by targeting disease pathogenesis. Curr. Treat. Options Rheumatol. 2017, 3, 33–48. [Google Scholar] [CrossRef]
- Kulkarni, H.S.; Kasi, P. Rituximab and cytokine release syndrome. Case Rep. Oncol. 2012, 5, 134–141. [Google Scholar] [CrossRef]
- Truffa-Bachi, P.; Lefkovits, I.; Frey, J.R. Proteomic analysis of T cell activation in the presence of cyclosporin A: Immunosuppressor and activator removal induces de novo protein synthesis. Mol. Immunol. 2000, 37, 261. [Google Scholar] [CrossRef]
- Caccavo, D.; Laganà, B.; Mitterhofer, A.P.; Ferri, G.M.; Afeltra, A.; Amoroso, A.; Bonomo, L. Long-term treatment of systemic lupus erythematosus with cyclosporin A. Arthritis Rheum. 1997, 40, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Summary of Product Characteristics, Labelling and Package Leaflet. Available online: Sandimmun-neoral-article-30-referral-annex-iii_en.pdf (accessed on 7 May 2020).
- Trottestam, H.; Horne, A.; Aricò, M.; Egeler, R.M.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Ladisch, S.; Webb, D.; Janka, G.; et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 treatment protocol. Blood 2011, 118, 4577–4584. [Google Scholar] [CrossRef] [PubMed]
- Moreland, L.W.; Bate, G.; Kirkpatrick, P. Abatacept. Nat. Rev. Drug Discov. 2006, 5, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Picchianti-Diamanti, A.; Rosado, M.; Germano, V.; Scarsella, M.; Giorda, E.; Podestà, E.; D’Amelio, R.; Carsetti, R.; Laganà, B. Reversion of resistance to immunosuppressive agents in three patients with psoriatic arthritis by cyclosporine A: Modulation of P-glycoprotein function. Clin. Immunol. 2011, 138, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, A.P.; Rosado, M.M.; Scarsella, M.; Germano, V.; Giorda, E.; Cascioli, S.; Lagana, B.; D’Amelio, R.; Carsetti, R. Abatacept (cytotoxic T lymphocyte antigen 4-immunoglobulin) improves B cell function and regulatory T cell inhibitory capacity in rheumatoid arthritis patients non-responding to anti-tumour necrosis factor-α agents. Clin. Exp. Immunol. 2014, 177, 630–640. [Google Scholar] [CrossRef]
- Mera-Varela, A.; Pérez-Pampín, E. Abatacept therapy in rheumatoid arthritis with interstitial lung disease. J. Clin. Rheumatol. 2014, 20, 445–446. [Google Scholar] [CrossRef]
- Record, J.L.; Beukelman, T.; Cron, R.Q.; Hasegawa, M.; Segawa, T.; Maeda, M.; Yoshida, T.; Sudo, A. Combination therapy of abatacept and anakinra in children with refractory systemic juvenile idiopathic arthritis: A retrospective case series. J. Rheumatol. 2011, 38, 180–181. [Google Scholar] [CrossRef]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 76. [Google Scholar] [CrossRef]
- Xiong, P.; Zeng, X.; Song, M.S.; Jia, S.W.; Zhong, M.H.; Xiao, L.L.; Lan, W.; Cai, C.; Wu, X.W.; Gong, F.L.; et al. Lack of association between Hla-A, -B and -DRB1 alleles and the development of SARS: A cohort of 95 SARS-recovered individuals in a population of Guangdong, Southern China. Int. J. Immunogenet. 2008, 35, 69–74. [Google Scholar] [CrossRef]
- Hajeer, A.H.; Balkhy, H.; Johani, S.; Yousef, M.Z.; Arabi, Y. Association of human leukocyte antigen class II alleles with severe middle east respiratory syndrome coronavirus infection. Ann. Thorac. Med. 2016, 11, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Glück, T.; Müller-Ladner, U. Vaccines: Vaccination in patients with chronic rheumatic or autoimmune diseases. Clin. Infect. Dis. 2008, 46, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L. Infections and biologic therapy in rheumatoid arthritis. Rheum. Dis. Clin. N. Am. 2012, 38, 727–745. [Google Scholar] [CrossRef] [PubMed]
- Smitten, A.L.; Choi, H.K.; Hochberg, M.C.; Suissa, S.; Simon, T.A.; Testa, M.A.; Chan, K. The risk of hospitalized infection in patients with rheumatoid arthritis. J. Rheumatol. 2008, 35, 387–393. [Google Scholar]
- Listing, J.; Gerhold, K.; Zink, A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology 2012, 52, 53–61. [Google Scholar] [CrossRef]
- Ramiro, S.; Sepriano, A.; Chatzidionysiou, K.; Nam, J.L.; Smolen, J.S.; van der Heijde, D.; Dougados, M.; van Vollenhoven, R.; Bijlsma, J.W.; Burmester, G.R.; et al. Safety of synthetic and biological DMARDs: A systematic literature review informing the 2016 update of the EULAR recommendations for management of rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1101–1136. [Google Scholar] [CrossRef]
- Germano, V.; Cattaruzza, M.S.; Osborn, J.; Tarantino, A.; Di Rosa, R.; Salemi, S.; D’Amelio, R. Infection risk in Rheumatoid Arthritis and Spondyloarthropathy patients under treatment with DMARDs, Corticosteroids and TNF-α antagonists. J. Transl. Med. 2014, 12, 77. [Google Scholar] [CrossRef]
- Au, K.; Reed, G.W.; Curtis, J.R.; Kremer, J.M.; Greenberg, J.D.; Strand, V.; Furst, D.E. High disease activity is associated with an increased risk of infection in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 785–791. [Google Scholar] [CrossRef]
- Dixon, W.G.; Suissa, S.; Hudson, M. The association between systemic glucocorticoid therapy and the risk of infection in patients with rheumatoid arthritis: Systematic review and meta-analyses. Arthritis Res. Ther. 2011, 13, R139. [Google Scholar] [CrossRef]
- Atzeni, F.; Sarzi-Puttini, P.; Botsios, C.; Carletto, A.; Cipriani, P.; Favalli, E.G.; Frati, E.; Foschi, V.; Gasparini, S.; Giardina, A.; et al. Long-term anti-TNF therapy and the risk of serious infections in a cohort of patients with rheumatoid arthritis: Comparison of adalimumab, etanercept and infliximab in the GISEA registry. Autoimmun. Rev. 2012, 12, 225–229. [Google Scholar] [CrossRef]
- Atzeni, F.; Sarzi-Puttini, P.; Mutti, A.; Bugatti, S.; Cavagna, L.; Caporali, R. Long-term safety of abatacept in patients with rheumatoid arthritis. Autoimmun. Rev. 2013, 12, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Wells, G.A.; Christensen, R.; Ghogomu, E.T.; Maxwell, L.J.; MacDonald, J.K.; Filippini, G.; Skoetz, N.; Francis, D.K.; Lopes, L.C.; et al. Adverse effects of biologics: A network meta-analysis and Cochrane overview. Cochrane Database Syst. Rev. 2011, 2, CD008794. [Google Scholar] [CrossRef] [PubMed]
- Holvast, A.; Huckriede, A.; Kallenberg, C.; Bijl, M. Influenza vaccination in systemic lupus erythematosus: Safe and protective? Autoimmun. Rev. 2007, 6, 300–305. [Google Scholar] [CrossRef]
- Kanterman, J.; Sade-Feldman, M.; Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 2012, 22, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Pandemia da COVID-19: la SIR Risponde ad Alcune Domande dei Pazienti—Società Italiana di Reumatologia. Available online: https://www.reumatologia.it/cmsx.asp?IDPg=1087 (accessed on 12 April 2020).
- Furer, V.; Rondaan, C.; Heijstek, M.W.; Levin, N.; Van Assen, S.; Bijl, M.; Breedveld, F.C.; D’Amelio, R.; Dougados, M.; Kapetanovic, M.C.; et al. 2019 update of EULAR recommendations for vaccination in adult patients with autoimmune inflammatory rheumatic diseases. Ann. Rheum. Dis. 2019, 79, 39–52. [Google Scholar] [CrossRef] [PubMed]


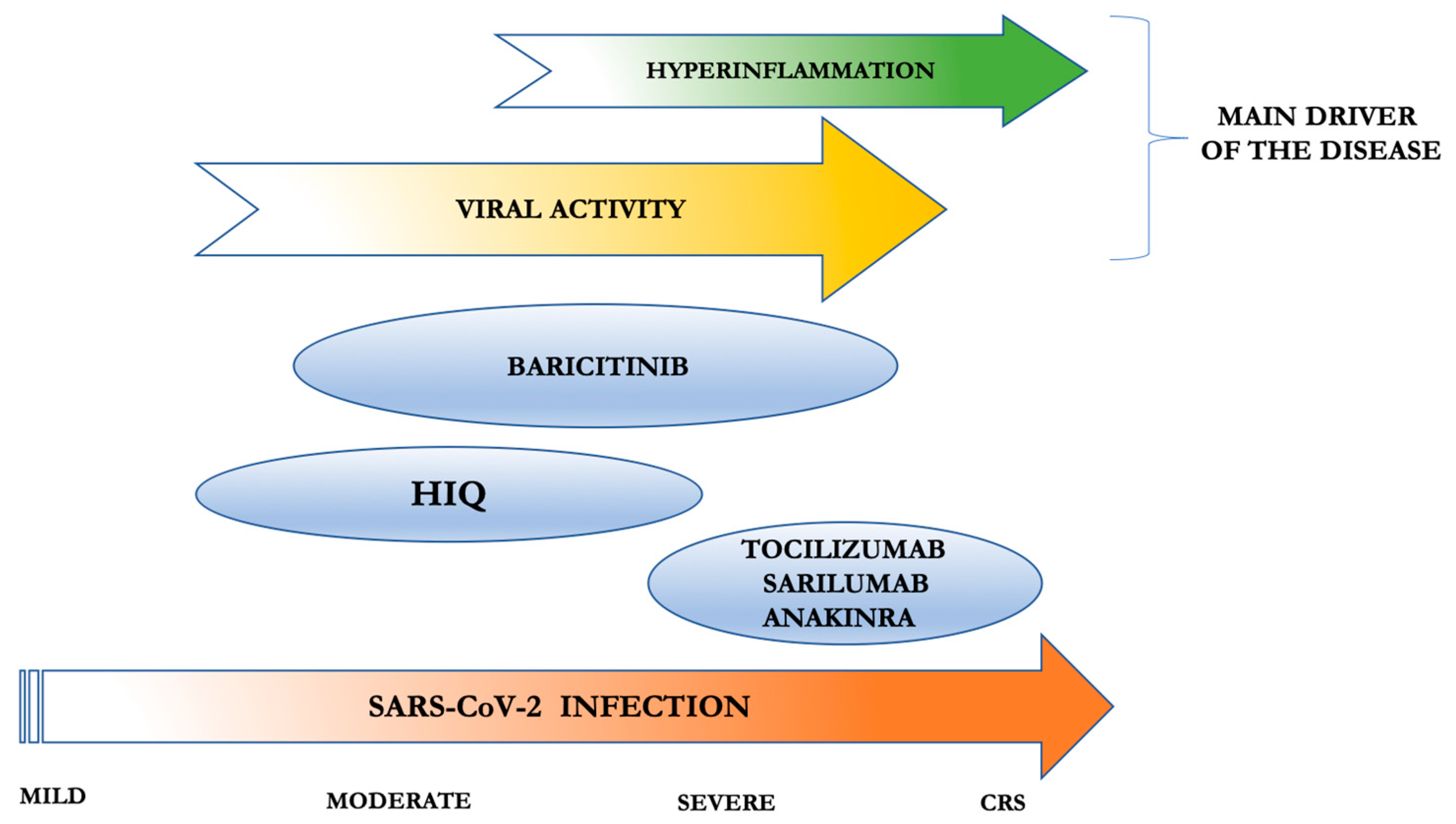{kind=link}
| Hydroxychloroquine/Chloroquine | Tocilizumab/Sarilumab | Anakinra | Baricitinib | Adalimumab | |
|---|---|---|---|---|---|
| Mechanism of Action | Immunomodulation = Impairing lysosomal functions Antiviral = Increasing endosomal pH and interfering with cellular receptors | Immunomodulation = IL-6 inhibition | Immunomodulation = IL-1 inhibition | Immunomodulation = IL-6 and IFN-γ inhibition Antiviral = AAK1 inhibition | Immunomodulation = TNF-α inhibition |
| Target Population | Mild with comorbidity Moderate/severe | Moderate/severe with or without CRS | Severe and CRS | Mild to severe, with or without CRS | Severe and CRS |
| Main Therapeutic Regimen | HIQ = 200–400 mg/day/orally CQ = 300 mg × 2/day/ orally | Single infusion 8 mg/kg/i.v. (max 800 mg) Second cycle after 8–12 h if no significant response ** | 100 mg/day/sc or 100 mg/4 times a day/i.v. | 2–4 mg/day/orally | Not reported |
| Main Safety Exclusion Criteria | Consider drug-to-drug interactions (i.e., QT interval prolongation) Retinopathy Severe renal dysfunction | Active TB and infections other than COVID-19 Bowel diverticulitis Severe heart failure Neut < 500/mmc Plt < 50,000/mmc Pregnancy | Active TB and infections other than COVID-19 NYHA class III/IV Severe renal dysfunction Pregnancy | Active TB and infections other than COVID-19 History of thrombophlebitis Severe renal dysfunction Pregnancy | Active TB and infections other than COVID-19 NYHA class III/IV Severe renal dysfunction Pregnancy |
| Specific Parameters to Closely Monitor | Blood count (reduction in Neut and Plt), AST, ALT, procalcitonin *, IL-6 | Blood count, AST, ALT, procalcitonin * | Blood count, AST, ALT, procalcitonin * | Blood count, AST, ALT, procalcitonin * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picchianti Diamanti, A.; Rosado, M.M.; Pioli, C.; Sesti, G.; Laganà, B. Cytokine Release Syndrome in COVID-19 Patients, A New Scenario for an Old Concern: The Fragile Balance between Infections and Autoimmunity. Int. J. Mol. Sci. 2020, 21, 3330. https://doi.org/10.3390/ijms21093330
Picchianti Diamanti A, Rosado MM, Pioli C, Sesti G, Laganà B. Cytokine Release Syndrome in COVID-19 Patients, A New Scenario for an Old Concern: The Fragile Balance between Infections and Autoimmunity. International Journal of Molecular Sciences. 2020; 21(9):3330. https://doi.org/10.3390/ijms21093330
Chicago/Turabian StylePicchianti Diamanti, Andrea, Maria Manuela Rosado, Claudio Pioli, Giorgio Sesti, and Bruno Laganà. 2020. "Cytokine Release Syndrome in COVID-19 Patients, A New Scenario for an Old Concern: The Fragile Balance between Infections and Autoimmunity" International Journal of Molecular Sciences 21, no. 9: 3330. https://doi.org/10.3390/ijms21093330
APA StylePicchianti Diamanti, A., Rosado, M. M., Pioli, C., Sesti, G., & Laganà, B. (2020). Cytokine Release Syndrome in COVID-19 Patients, A New Scenario for an Old Concern: The Fragile Balance between Infections and Autoimmunity. International Journal of Molecular Sciences, 21(9), 3330. https://doi.org/10.3390/ijms21093330