Toll-Like Receptor Signaling and Immune Regulatory Lymphocytes in Periodontal Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Toll-Like Receptor (TLR) Signaling in the Etiology of Periodontitis
3. Immune T and B Cells in Periodontitis Pathogenesis
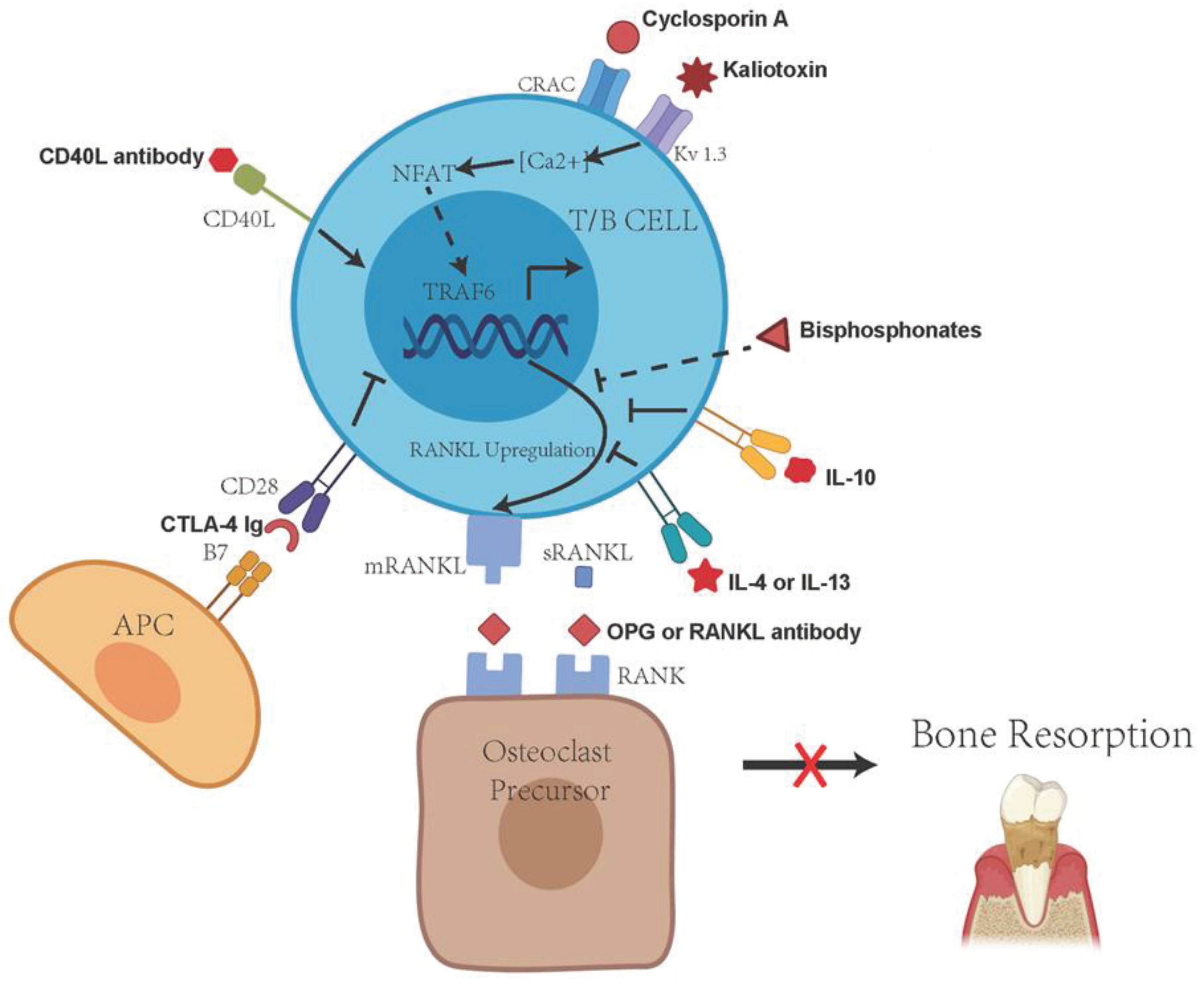
4. Immune Cell RANKL Activity in the Progression and Pathogenesis of Periodontitis
5. TLR Signaling and Regulation of RANKL Activity
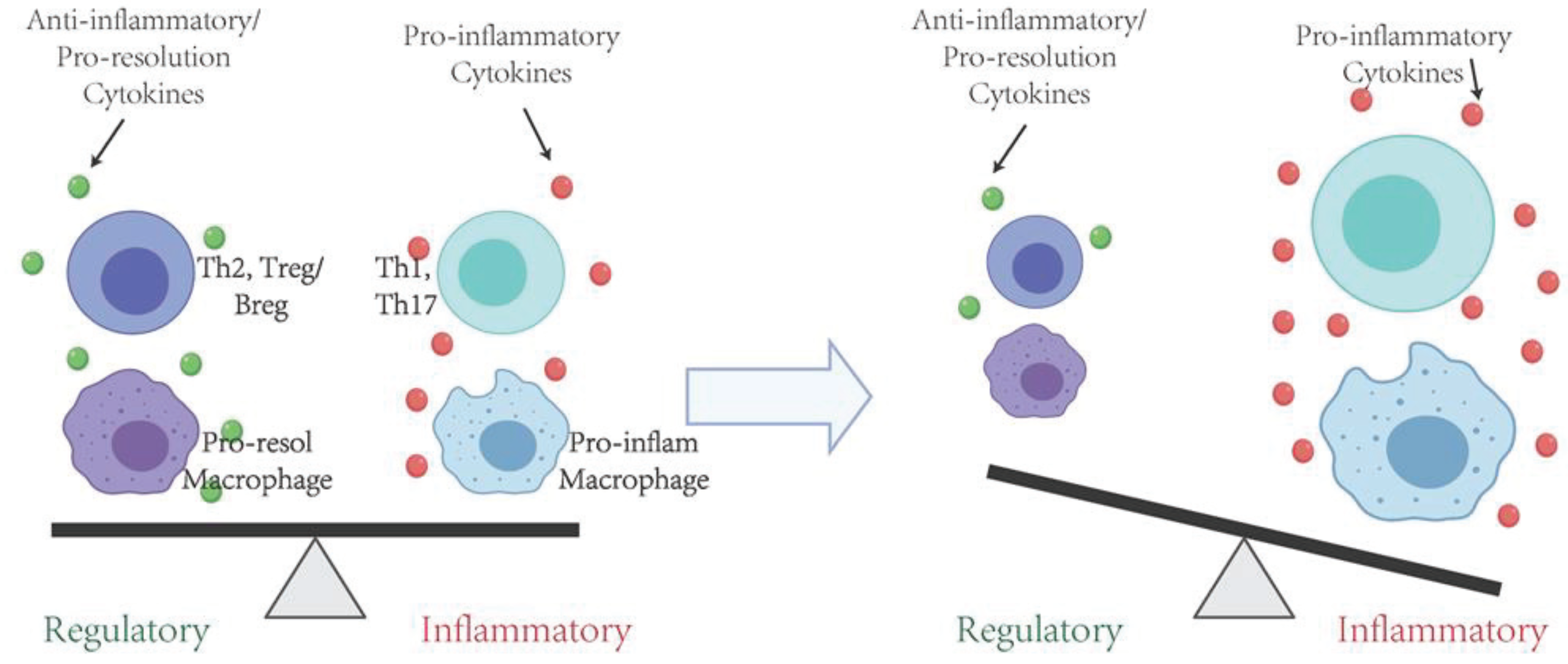
6. Protective Effects of Immune Regulatory T Cells in the Inflammation and Periodontal Bone Resorption
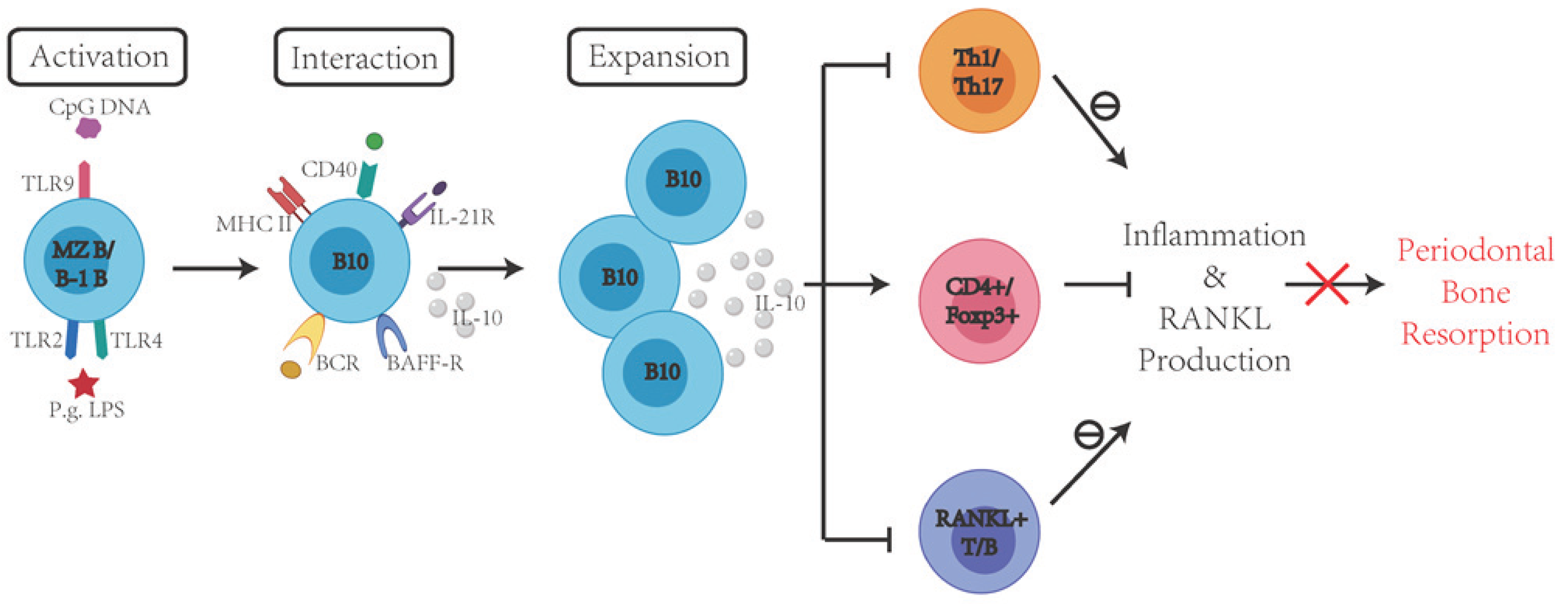
7. Protective Effects of Immune Regulatory B Cells in the Inflammation and Periodontal Bone Resorption
8. Concluding Remarks
Funding
Conflicts of Interest
References
- Herrera, D.; Retamal-Valdes, B.; Alonso, B.; Feres, M. Acute periodontal lesions (periodontal abscesses and necrotizing periodontal diseases) and endo-periodontal lesions. J. Periodontol. 2018, 89, S85–S102. [Google Scholar] [CrossRef]
- Albandar, J.M.; Susin, C.; Hughes, F.J. Manifestations of systemic diseases and conditions that affect the periodontal attachment apparatus: Case definitions and diagnostic considerations. J. Periodontol. 2018, 89, S183–S203. [Google Scholar] [CrossRef]
- Needleman, I.; Garcia, R.; Gkranias, N.; Kirkwood, K.L.; Kocher, T.; Iorio, A.D.; Moreno, F.; Petrie, A. Mean annual attachment, bone level, and tooth loss: A systematic review. J. Periodontol. 2018, 89, S120–S139. [Google Scholar] [CrossRef]
- Billings, M.; Holtfreter, B.; Papapanou, P.N.; Mitnik, G.L.; Kocher, T.; Dye, B.A. Age-dependent distribution of periodontitis in two countries: Findings from NHANES 2009 to 2014 and SHIP-TREND 2008 to 2012. J. Clin. Periodontol. 2018, 45, S130–S148. [Google Scholar] [CrossRef]
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89, S173–S182. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Patil, A.G.; Loos, B.G. Classification and diagnosis of aggressive periodontitis. J. Periodontol. 2018, 89, S103–S119. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.S.; Greenwell, H.; Kornman, K.S. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J. Periodontol. 2018, 89, S159–S172. [Google Scholar] [CrossRef] [PubMed]
- Mombelli, A. Microbial colonization of the periodontal pocket and its significance for periodontal therapy. Periodontol. 2000 2017, 76, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Sochalska, M.; Potempa, J. Manipulation of Neutrophils by Porphyromonas gingivalis in the Development of Periodontitis. Front. Microbiol. 2017, 7, 197. [Google Scholar] [CrossRef]
- Akrivopoulou, C.; Green, I.; Donos, N.; Nair, S.P.; Ready, D. Aggregatibacter actinomycetemcomitans serotype prevalence and antibiotic resistance in a UK population with periodontitis. J. Glob. Antimicrob. Resist. 2017, 10, 54–58. [Google Scholar] [CrossRef]
- Mahalakshmi, K.; Krishnan, P.; Chandrasekaran, S. Detection of Tannerella forsythia bspA and prtH genotypes among periodontitis patients and healthy subjects—A case—Control study. Arch. Oral Boil. 2018, 96, 178–181. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, K.Q.; Da Silva, C.L.C.A.; Coutinho-Silva, R. Immunological Pathways Triggered byPorphyromonas gingivalisandFusobacterium nucleatum: Therapeutic Possibilities? Mediat. Inflamm. 2019, 2019, 7241312. [Google Scholar] [CrossRef] [PubMed]
- Kokubu, E.; Inoue, T.; Ishihara, K. Response of epithelial cells infected by Treponema denticola. Oral Dis. 2018, 24, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Willmann, C.; Mata, X.; Hanghøj, K.; Tonasso, L.; Tisseyre, L.; Jeziorski, C.; Cabot, É.; Chevet, P.; Crubézy, É.; Orlando, L.; et al. Oral health status in historic population: Macroscopic and metagenomic evidence. PLoS ONE 2018, 13, e0196482. [Google Scholar] [CrossRef]
- Yumoto, H.; Hirota, K.; Hirao, K.; Miyazaki, T.; Yamamoto, N.; Miyamoto, K.; Murakami, K.; Fujiwara, N.; Matsuo, T.; Miyake, Y. Anti-inflammatory and protective effects of 2-methacryloyloxyethyl phosphorylcholine polymer on oral epithelial cells. J. Biomed. Mater. Res. Part A 2014, 103, 555–563. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Lee, M.K.; Singleton, W.; Achuthan, A.; Lee, M.-C.; O’Brien-Simpson, N.M.; Cook, A.D.; Murphy, A.J.; Dashper, S.G.; Reynolds, E.C.; et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Front. Microbiol. 2017, 7, 351. [Google Scholar] [CrossRef]
- Gholizadeh, P.; Pormohammad, A.; Eslami, H.; Shokouhi, B.; Fakhrzadeh, V.; Kafil, H.S. Oral pathogenesis of Aggregatibacter actinomycetemcomitans. Microb. Pathog. 2017, 113, 303–311. [Google Scholar] [CrossRef]
- Groeger, S.E.; Meyle, J. Epithelial barrier and oral bacterial infection. Periodontol. 2000 2015, 69, 46–67. [Google Scholar] [CrossRef]
- Cueno, M.E.; Ochiai, K. Gingival Periodontal Disease (PD) Level-Butyric Acid Affects the Systemic Blood and Brain Organ: Insights Into the Systemic Inflammation of Periodontal Disease. Front. Immunol. 2018, 9, 1158. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Ho, Y.-P.; Chou, Y.-S.; Ho, K.-Y.; Wu, Y.-M.; Lin, Y.-C. Aggregatibacter (Actinobacillus) actimycetemcomitans leukotoxin and human periodontitis—A historic review with emphasis on JP2. Kaohsiung J. Med. Sci. 2018, 34, 186–193. [Google Scholar] [CrossRef]
- Greabu, M.; Totan, A.; Miricescu, D.; Radulescu, R.; Virlan, J.; Calenic, B. Hydrogen Sulfide, Oxidative Stress and Periodontal Diseases: A Concise Review. Antioxidants 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Meyle, J.; Dommisch, H.; Groeger, S.; Giacaman, R.A.; Costalonga, M.; Herzberg, M. The innate host response in caries and periodontitis. J. Clin. Periodontol. 2017, 44, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Taubman, M.A.; Yoshie, H.; Ebersole, J.; Smith, D.; Olson, C. Host Response in Experimental Periodontal Disease. J. Dent. Res. 1984, 63, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Wang, Q.; Chen, Q. The cytokine network involved in the host immune response to periodontitis. Int. J. Oral Sci. 2019, 11, 30. [Google Scholar] [CrossRef]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune Response: The Key to Bone Resorption in Periodontal Disease. J. Periodontol. 2005, 76, 2033–2041. [Google Scholar] [CrossRef]
- Lundmark, A.; Hu, Y.O.O.; Huss, M.; Johannsen, G.; Andersson, A.F.; Yucel-Lindberg, T. Identification of Salivary Microbiota and Its Association With Host Inflammatory Mediators in Periodontitis. Front. Microbiol. 2019, 9, 216. [Google Scholar] [CrossRef]
- Taubman, M.A.; Kawai, T. Involvement of T-lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit. Rev. Oral Boil. Med. 2001, 12, 125–135. [Google Scholar] [CrossRef]
- Shaddox, L.M.; Spencer, W.P.; Velsko, I.M.; Al-Kassab, H.; Huang, H.; Calderon, N.; Aukhil, I.; Wallet, S.M.; Hiba, H. Localized aggressive periodontitis immune response to healthy and diseased subgingival plaque. J. Clin. Periodontol. 2016, 43, 746–753. [Google Scholar] [CrossRef]
- Naruishi, K.; Nagata, T. Biological effects of interleukin-6 on Gingival Fibroblasts: Cytokine regulation in periodontitis. J. Cell. Physiol. 2018, 233, 6393–6400. [Google Scholar] [CrossRef]
- Cavalla, F.; Osorio, C.; Paredes, R.; Valenzuela, M.A.; Garcia-Sesnich, J.; Sorsa, T.; Tervahartiala, T.; Hernández, M. Matrix metalloproteinases regulate extracellular levels of SDF-1/CXCL12, IL-6 and VEGF in hydrogen peroxide-stimulated human periodontal ligament fibroblasts. Cytokine 2015, 73, 114–121. [Google Scholar] [CrossRef]
- Han, M.-X.; Ding, C.; Kyung, H.M. Genetic polymorphisms in pattern recognition receptors and risk of periodontitis: Evidence based on 12,793 subjects. Hum. Immunol. 2015, 76, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X. Lipopolysaccharide-regulated production of bone sialoprotein and interleukin-8 in human periodontal ligament fibroblasts: The role of toll-like receptors 2 and 4 and the MAPK pathway. J. Periodontal Res. 2014, 50, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Alqallaf, H.; Hamada, Y.; Blanchard, S.; Shin, D.; Gregory, R.; Srinivasan, M. Differential profiles of soluble and cellular toll like receptor (TLR)-2 and 4 in chronic periodontitis. PLoS ONE 2018, 13, e0200231. [Google Scholar] [CrossRef]
- Ilango, P.; Mahalingam, A.; Parthasarathy, H.; Katamreddy, V.; Subbareddy, V. Evaluation of TLR2 and 4 in Chronic Periodontitis. J. Clin. Diagn. Res. 2016, 10, ZC86–ZC89. [Google Scholar] [CrossRef]
- Babu, S.; Blauvelt, C.P.; Kumaraswami, V.; Nutman, T.B. Cutting edge: Diminished T cell TLR expression and function modulates the immune response in human filarial infection. J. Immunol. 2006, 176, 3885–3889. [Google Scholar] [CrossRef]
- Dorner, M.; Brandt, S.; Tinguely, M.; Zucol, F.; Bourquin, J.-P.; Zauner, L.; Berger, C.; Bernasconi, M.; Speck, R.F.; Nadal, D. Plasma cell toll-like receptor (TLR) expression differs from that of B cells, and plasma cell TLR triggering enhances immunoglobulin production. Immunol. 2009, 128, 573–579. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, Y.; Yang, Q.; Wang, S.; Zou, B.; Tan, Y.; Zou, M.; Liu, S.; Li, X. Artesunate attenuates LPS-induced osteoclastogenesis by suppressing TLR4/TRAF6 and PLCγ1-Ca2+-NFATc1 signaling pathway. Acta Pharmacol. Sin. 2020, 41, 229–236. [Google Scholar] [CrossRef]
- Li, K.; Lv, G.; Pan, L. Sirt1 alleviates LPS induced inflammation of periodontal ligament fibroblasts via downregulation of TLR4. Int. J. Boil. Macromol. 2018, 119, 249–254. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.-C.; Lu, W.; Song, Z.-C.; Zhou, W. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflammation 2018, 15, 37. [Google Scholar] [CrossRef]
- Ding, P.-H.; Darveau, R.P.; Wang, C.-Y.; Jin, L. 3LPS-binding protein and its interactions with P. gingivalis LPS modulate pro-inflammatory response and Toll-like receptor signaling in human oral keratinocytes. PLoS ONE 2017, 12, e0173223. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, S.S.; Velsko, I.M.; Rivera-Kweh, M.F.; Larjava, H.; Lucas, A.; Kesavalu, L. Global TLR2 and 4 deficiency in mice impacts bone resorption, inflammatory markers and atherosclerosis to polymicrobial infection. Mol. Oral Microbiol. 2016, 32, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Sojar, H.; Genco, R.J.; DeNardin, E. Intracellular Signaling and Cytokine Induction upon Interactions ofPorphyromonas gingivalisFimbriae with Pattern-Recognition Receptors. Immunol. Investig. 2004, 33, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.D.; Xia-Juan, X.; Crump, K.E.; Abe, T.; Hajishengallis, G.; Sahingur, S.E. Toll-Like Receptor 9-Mediated Inflammation Triggers Alveolar Bone Loss in Experimental Murine Periodontitis. Infect. Immun. 2015, 83, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Zhang, Y.; Chen, L.; Zhou, T.; Huang, W.; Zhou, X.; Shao, L.; Zhang, Y.; Chen, L.; Huang, W.; et al. The role of Toll-like receptors in periodontitis. Oral Dis. 2016, 23, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Crump, K.E.; Oakley, J.C.; Xia-Juan, X.; Madu, T.C.; Devaki, S.; Mooney, E.C.; Sahingur, S.E. Interplay of Toll-Like Receptor 9, Myeloid Cells, and Deubiquitinase A20 in Periodontal Inflammation. Infect. Immun. 2016, 85, e00814-16. [Google Scholar] [CrossRef]
- Souto, G.R.; Queiroz, C.M.; Costa, F.O.; Mesquita, R.A. Relationship Between Chemokines and Dendritic Cells in Human Chronic Periodontitis. J. Periodontol. 2014, 85, 1416–1423. [Google Scholar] [CrossRef]
- Cavalla, F.; Araujo-Pires, A.C.; Biguetti, C.C.; Garlet, G.P. Cytokine Networks Regulating Inflammation and Immune Defense in the Oral Cavity. Curr. Oral Health Rep. 2014, 1, 104–113. [Google Scholar] [CrossRef]
- Fiorillo, L.; Cervino, G.; Herford, A.; Lauritano, F.; D’Amico, C.; Giudice, R.L.; Laino, L.; Troiano, G.; Crimi, S.; Cicciù, M. Interferon Crevicular Fluid Profile and Correlation with Periodontal Disease and Wound Healing: A Systemic Review of Recent Data. Int. J. Mol. Sci. 2018, 19, 1908. [Google Scholar] [CrossRef]
- Silva, N.; Abusleme, L.; Bravo, D.; Dutzan, N.; Garcia-Sesnich, J.; Vernal, R.; Hernández, M.; Gamonal, J. Host response mechanisms in periodontal diseases. J. Appl. Oral Sci. 2015, 23, 329–355. [Google Scholar] [CrossRef]
- Boelen, G.-J.; Boute, L.; D’Hoop, J.; Ezeldeen, M.; Lambrichts, I.; Opdenakker, G. Matrix metalloproteinases and inhibitors in dentistry. Clin. Oral Investig. 2019, 23, 2823–2835. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Hu, Z.; Ge, S. Healthy and Inflamed Gingival Fibroblasts Differ in Their Inflammatory Response to Porphyromonas gingivalis Lipopolysaccharide. Inflammation 2016, 39, 1842–1852. [Google Scholar] [CrossRef]
- Ohgi, K.; Kajiya, H.; Goto-T, K.; Okamoto, F.; Yoshinaga, Y.; Okabe, K.; Sakagami, R. Toll-like receptor 2 activation primes and upregulates osteoclastogenesis via lox-1. Lipids Health Dis. 2018, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Kaneko, T.; Ukai, T.; Yokoyama, M.; Haro, E.A.; Yoshinaga, Y.; Yoshimura, A.; Hara, Y. Peptidoglycan and lipopolysaccharide synergistically enhance bone resorption and osteoclastogenesis. J. Periodontal Res. 2012, 47, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, J.; Xu, Q.; Harber, G.; Feng, X.; Michalek, S.M.; Katz, J. TLR2-dependent modulation of osteoclastogenesis by Porphyromonas gingivalis through differential induction of NFATc1 and NF-kappaB. J. Biol. Chem. 2011, 286, 24159–24169. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hu, Y.; Freire, M.; Yu, P.; Kawai, T.; Han, X. Role of toll-like receptor 2 in inflammation and alveolar bone loss in experimental peri-implantitis versus periodontitis. J. Periodontal Res. 2017, 53, 98–106. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone–immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef]
- Lin, X.; Han, X.; Kawai, T.; Taubman, M.A. Antibody to Receptor Activator of NF-κB Ligand Ameliorates T Cell-Mediated Periodontal Bone Resorption ▿. Infect. Immun. 2010, 79, 911–917. [Google Scholar] [CrossRef]
- Gruber, R. Osteoimmunology: Inflammatory osteolysis and regeneration of the alveolar bone. J. Clin. Periodontol. 2019, 46, 52–69. [Google Scholar] [CrossRef]
- Han, X.; Kawai, T.; Taubman, M.A. Interference with immune-cell-mediated bone resorption in periodontal disease. Periodontol. 2000 2007, 45, 76–94. [Google Scholar] [CrossRef]
- Li, S.; Hao, L.; Wang, L.; Lu, Y.; Li, Q.; Zhu, Z.; Shao, J.-Z.; Chen, W. Targeting Atp6v1c1 Prevents Inflammation and Bone Erosion Caused by Periodontitis and Reveals Its Critical Function in Osteoimmunology. PLoS ONE 2015, 10, e0134903. [Google Scholar] [CrossRef] [PubMed]
- Parente, R.; Sobacchi, C.; Bottazzi, B.; Mantovani, A.; Grčevic, D.; Inforzato, A. The Long Pentraxin PTX3 in Bone Homeostasis and Pathology. Front. Immunol. 2019, 10, 2628. [Google Scholar] [CrossRef] [PubMed]
- Sima, C.; Viniegra, A.; Glogauer, M. Macrophage immunomodulation in chronic osteolytic diseases-the case of periodontitis. J. Leukoc. Boil. 2018, 105, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Taubman, M.A.; Kawai, T.; Han, X. The new concept of periodontal disease pathogenesis requires new and novel therapeutic strategies. J. Clin. Periodontol. 2007, 34, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, E.M.; Arosa, F. CD8+ T Cells in Chronic Periodontitis: Roles and Rules. Front. Immunol. 2017, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.-T.A. The role of acquired immunity and periodontal disease progression. Crit. Rev. Oral Boil. Med. 2003, 14, 237–252. [Google Scholar] [CrossRef]
- Han, X.; Lin, X.; Yu, X.; Lin, J.; Kawai, T.; LaRosa, K.B.; Taubman, M.A. Porphyromonas gingivalis Infection-Associated Periodontal Bone Resorption Is Dependent on Receptor Activator of NF-κB Ligand. Infect. Immun. 2013, 81, 1502–1509. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef]
- Yoshie, H.; Taubman, M.A.; Olson, C.L.; Ebersole, J.L.; Smith, D.J.; Yoshie, H. Periodontal bone loss and immune characteristics after adoptive transfer of Actinobacilus-sensitized T cells to rats. J. Periodontal Res. 1987, 22, 499–505. [Google Scholar] [CrossRef]
- Baker, P.J.; Garneau, J.; Howe, L.; Roopenian, D.C. T-cell contributions to alveolar bone loss in response to oral infection withPorphyromonas gingivalis. Acta Odontol. Scand. 2001, 59, 222–225. [Google Scholar] [CrossRef]
- Kawai, T.; Shimauchi, H.; Eastcott, J.W.; Smith, D.J.; Taubman, M.A. Antigen direction of specific T-cell clones into gingival tissues. Immunology 1998, 93, 11–19. [Google Scholar] [CrossRef]
- Najafian, N.; Sayegh, M.H. CTLA4-Ig: A novel immunosuppressive agent. Expert Opin. Investig. Drugs 2000, 9, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, E.; McHugh, G.B.; Grieco, D.A.; Seymour, G. Costimulatory molecules in human periodontal disease tissues. J. Periodontal Res. 2001, 36, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Eastcott, J.W.; Taubman, M.A.; Smith, D.J.; Cox, D.S. Effect of adoptive transfer of cloned Actinobacillus actinomycetemcomitans-specific T helper cells on periodontal disease. Infect. Immun. 1991, 59, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Eastcott, J.W.; Yamashita, K.; Taubman, M.A.; Smith, D.J. Characterization of rat T-cell clones with bacterial specificity. Immunology 1990, 71, 120–126. [Google Scholar]
- Weitzmann, M.N. Bone and the Immune System. Toxicol. Pathol. 2017, 45, 911–924. [Google Scholar] [CrossRef]
- Kozuka, Y.; Ozaki, Y.; Ukai, T.; Kaneko, T.; Hara, Y. B Cells Play an Important Role in Lipopolysaccharide-Induced Bone Resorption. Calcif. Tissue Int. 2006, 78, 125–132. [Google Scholar] [CrossRef]
- Han, X.; Kawai, T.; Eastcott, J.W.; Taubman, M.A. Bacterial-responsive B lymphocytes induce periodontal bone resorption. J. Immunol. 2006, 176, 625–631. [Google Scholar] [CrossRef]
- Harada, Y.; Han, X.; Yamashita, K.; Kawai, T.; Eastcott, J.W.; Smith, D.J.; Taubman, M.A. Effect of adoptive transfer of antigen-specific B cells on periodontal bone resorption. J. Periodontal Res. 2006, 41, 101–107. [Google Scholar] [CrossRef]
- Kawai, T.; Eisen-Lev, R.; Seki, M.; Eastcott, J.W.; Wilson, M.E.; Taubman, M.A. Requirement of B7 costimulation for Th1-mediated inflammatory bone resorption in experimental periodontal disease. J. Immunol. 2000, 164, 2102–2109. [Google Scholar] [CrossRef]
- Kenkre, J.; Bassett, J.H.D. The bone remodelling cycle. Ann. Clin. Biochem. Int. J. Lab. Med. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J.; Sims, N. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-Y.; Kim, Y.-K. Berberine Suppresses RANKL-Induced Osteoclast Differentiation by Inhibiting c-Fos and NFATc1 Expression. Am. J. Chin. Med. 2019, 47, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, H.; Makihira, S.; Suzuki, M.; Ishii, T.; Movila, A.; Hirschfeld, J.; Mawardi, H.; Lin, X.; Han, X.; Taubman, M.A.; et al. Soluble RANKL Cleaved from Activated Lymphocytes by TNF-α-Converting Enzyme Contributes to Osteoclastogenesis in Periodontitis. J. Immunol. 2016, 197, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, A.; Kanno, T.; Hoshi, M.; Shibata, O.; Yano, K.; Fujise, N.; Kinosaki, M.; Yamaguchi, K.; Tsuda, E.; Murakami, A.; et al. Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J. Bone Miner. Metab. 2002, 20, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Xiao, F.; Yuan, K.; Hu, X.; Lin, W.; Ma, R.; Zhang, X.; Huang, Z. Nardosinone Suppresses RANKL-Induced Osteoclastogenesis and Attenuates Lipopolysaccharide-Induced Alveolar Bone Resorption. Front. Pharmacol. 2017, 8, 626. [Google Scholar] [CrossRef]
- Kuritani, M.; Sakai, N.; Karakawa, A.; Isawa, M.; Chatani, M.; Negishi-Koga, T.; Funatsu, T.; Takami, M. Anti-mouse RANKL Antibodies Inhibit Alveolar Bone Destruction in Periodontitis Model Mice. Boil. Pharm. Bull. 2018, 41, 637–643. [Google Scholar] [CrossRef]
- Degertekin, C.K.; Turhan Iyidir, O.; Yılmaz, B.A.; Elbeg, S.; Pasaoglu, O.T.; Pasaoglu, H.; Cakır, N.; Arslan, M. RANKL/Osteoprotegerin System and Bone Turnover in Hashimoto Thyroiditis. Calcif. Tissue Int. 2016, 99, 365–372. [Google Scholar] [CrossRef]
- Ferreira, E.C.; Bortolin, R.H.; Freire-Neto, F.P.; Souza, K.S.; Bezerra, J.; Ururahy, M.A.; Ramos, A.M.D.O.; Himelfarb, S.T.; Abreu, B.J.; DiDone, T.V.; et al. Zinc supplementation reduces RANKL/OPG ratio and prevents bone architecture alterations in ovariectomized and type 1 diabetic rats. Nutr. Res. 2017, 40, 48–56. [Google Scholar] [CrossRef]
- El Kholy, K.; Freire, M.; Chen, T.; Van Dyke, T.E. Resolvin E1 Promotes Bone Preservation Under Inflammatory Conditions. Front. Immunol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Costa, L.C.; Da Fonseca, M.A.; Pinheiro, A.D.R.; Aguiar, T.R.D.S.; Machado, A.N.; Quinelato, V.; Bonato, L.L.; Aguiar, D.P.; Vieira, T.; De Almeida, F.L.D.; et al. Chronic Periodontitis and RANKL/OPG Ratio in Peri-Implant Mucosae Inflammation. Braz. Dent. J. 2018, 29, 14–22. [Google Scholar] [CrossRef]
- Salinas-Muñoz, M.; Garrido-Flores, M.; Baeza, M.; Huaman, P.; García-Sesnich, J.; Bologna, R.; Vernal, R.; Hernández, M. Bone resorptive activity in symptomatic and asymptomatic apical lesions of endodontic origin. Clin. Oral Investig. 2017, 21, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, A.; Fukada, S.Y.; De Rossi, M.; Da Silva, R.A.B.; Queiroz, A.M.; Nelson-Filho, P.; Da Silva, L.A.B. Cementocytes Express Receptor Activator of the Nuclear Factor Kappa-B Ligand in Response to Endodontic Infection in Mice. J. Endod. 2016, 42, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M.; Looney, S.; Singh, N.; Elashiry, M.; Meghil, M.M.; El-Awady, A.R.; Tawfik, O.; Susin, C.; Arce, R.M.; Cutler, C.W. Systemic Antibiotic Therapy Reduces Circulating Inflammatory Dendritic Cells and Treg-Th17 Plasticity in Periodontitis. J. Immunol. 2019, 202, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Braz-Silva, P.H.; Bergamini, M.L.; Mardegan, A.P.; De Rosa, C.S.; Hasseus, B.; Jonasson, P. Inflammatory profile of chronic apical periodontitis: A literature review. Acta Odontol. Scand. 2018, 77, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Yuce, H.B.; Gokturk, O.; Turkal, H.A.; Inanir, A.; Benli, I.; Demir, O. Assessment of local and systemic 25-hydroxy-vitamin D, RANKL, OPG, and TNF levels in patients with rheumatoid arthritis and periodontitis. J. Oral Sci. 2017, 59, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, C.M.D.S.; Lira-Junior, R.; Love, R. T and B Cells in Periodontal Disease: New Functions in A Complex Scenario. Int. J. Mol. Sci. 2019, 20, 3949. [Google Scholar] [CrossRef]
- Melgar-Rodríguez, S.; Díaz-Zúñiga, J.; Alvarez, C.; Rojas, L.; Monasterio, G.; Carvajal, P.; Escobar, A.; Sanz, M.; Vernal, R. Serotype b ofAggregatibacter actinomycetemcomitansincreases osteoclast and memory T-lymphocyte activation. Mol. Oral Microbiol. 2015, 31, 162–174. [Google Scholar] [CrossRef]
- Palioto, D.B.; Finoti, L.S.; Kinane, D.F.; Benakanakere, M. Epigenetic and inflammatory events in experimental periodontitis following systemic microbial challenge. J. Clin. Periodontol. 2019, 46, 819–829. [Google Scholar] [CrossRef]
- Chen, B.; Wu, W.; Sun, W.; Zhang, Q.; Yan, F.; Xiao, Y. RANKL Expression in Periodontal Disease: Where Does RANKL Come from? BioMed Res. Int. 2014, 2014, 731039. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Jin, Y.; Miao, Y.; Shi, T.; Lin, X. Improved RANKL production by memory B cells: A way for B cells promote alveolar bone destruction during periodontitis. Int. Immunopharmacol. 2018, 64, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Jin, Y.; Miao, Y.; Shi, T.; Lin, X. Improved RANKL expression and osteoclastogenesis induction of CD27+CD38− memory B cells: A link between B cells and alveolar bone damage in periodontitis. J. Periodontal Res. 2019, 54, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Matsuyama, T.; Hosokawa, Y.; Makihira, S.; Seki, M.; Karimbux, N.Y.; Gonçalves, R.B.; Valverde, P.; Dibart, S.; Li, Y.-P.; et al. B and T Lymphocytes Are the Primary Sources of RANKL in the Bone Resorptive Lesion of Periodontal Disease. Am. J. Pathol. 2006, 169, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Nakayamada, S.; Kubo, S.; Nakano, K.; Iwata, S.; Miyagawa, I.; Ma, X.; Trimova, G.; Sakata, K.; Tanaka, Y. Th22 Cells Promote Osteoclast Differentiation via Production of IL-22 in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 2901. [Google Scholar] [CrossRef] [PubMed]
- Manabe, N.; Kawaguchi, H.; Chikuda, H.; Miyaura, C.; Inada, M.; Nagai, R.; Nabeshima, Y.-I.; Nakamura, K.; Sinclair, A.M.; Scheuermann, R.H.; et al. Connection between B lymphocyte and osteoclast differentiation pathways. J. Immunol. 2001, 167, 2625–2631. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.-T.A.; Nguyen, H.; Gao, X.; Kong, Y.-Y.; Gorczynski, R.M.; Singh, B.; Ellen, R.P.; Penninger, J.M. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J. Clin. Investig. 2000, 106, R59–R67. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Paster, B.J.; Komatsuzawa, H.; Ernst, C.W.O.; Goncalves, R.B.; Sasaki, H.; Ouhara, K.; Stashenko, P.P.; Sugai, M.; Taubman, M.A. Cross-reactive adaptive immune response to oral commensal bacteria results in an induction of receptor activator of nuclear factor-?B ligand (RANKL)-dependent periodontal bone resorption in a mouse model. Oral Microbiol. Immunol. 2007, 22, 208–215. [Google Scholar] [CrossRef]
- Crotti, T.; Smith, M.D.; Hirsch, R.; Soukoulis, S.; Weedon, H.; Capone, M.; Ahern, M.J.; Haynes, D. Receptor activator NF κB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J. Periodontal Res. 2003, 38, 380–387. [Google Scholar] [CrossRef]
- Jin, Q.; Cirelli, J.A.; Park, C.H.; Sugai, J.V.; Taba, M.; Kostenuik, P.J.; Giannobile, W. RANKL inhibition through osteoprotegerin blocks bone loss in experimental periodontitis. J. Periodontol. 2007, 78, 1300–1308. [Google Scholar] [CrossRef]
- Kassem, A.; Henning, P.; Lundberg, P.; De Souza, P.P.C.; Lindholm, C.; Lerner, U.H. Porphyromonas gingivalis Stimulates Bone Resorption by Enhancing RANKL (Receptor Activator of NF-κB Ligand) through Activation of Toll-like Receptor 2 in Osteoblasts*. J. Boil. Chem. 2015, 290, 20147–20158. [Google Scholar] [CrossRef]
- Amcheslavsky, A.; Bar-Shavit, Z. Interleukin (IL)-12 mediates the anti-osteoclastogenic activity of CpG-oligodeoxynucleotides. J. Cell. Physiol. 2006, 207, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.-D.; Park-Min, K.-H.; Shen, Z.; Fajardo, R.J.; Goldring, S.R.; McHugh, K.P.; Ivashkiv, L.B. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J. Immunol. 2009, 183, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lin, J.; Yu, Q.; Kawai, T.; Taubman, M.A.; Han, X. Activation of Toll-like receptor 9 inhibits lipopolysaccharide-induced receptor activator of nuclear factor kappa- B ligand expression in rat B lymphocytes. Microbiol. Immunol. 2014, 58, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Hu, Y.; Wang, Y.; Kawai, T.; Wang, Z.; Han, X. Different engagement of TLR2 and TLR4 in Porphyromonas gingivalis vs. ligature-induced periodontal bone loss. Braz. Oral Res. 2017, 31, e63. [Google Scholar] [CrossRef] [PubMed]
- AlQranei, M.S.; Chellaiah, M.A. Osteoclastogenesis in periodontal diseases: Possible mediators and mechanisms. J. Oral Biosci. 2020. [Google Scholar] [CrossRef]
- Garlet, G.P.; Sfeir, C.S.; Little, S.R. Restoring host-microbe homeostasis via selective chemoattraction of Tregs. J. Dent. Res. 2014, 93, 834–839. [Google Scholar] [CrossRef]
- Garlet, G.P.; Cardoso, C.R.D.B.; Mariano, F.S.; Claudino, M.; Assis, G.F.; Campanelli, A.P.; Avila-Campos, M.; Silva, J.S. Regulatory T cells attenuate experimental periodontitis progression in mice. J. Clin. Periodontol. 2009, 37, 591–600. [Google Scholar] [CrossRef]
- Glowacki, A.J.; Gottardi, R.; Yoshizawa, S.; Cavalla, F.; Garlet, G.P.; Sfeir, C.; Little, S.R. Strategies to direct the enrichment, expansion, and recruitment of regulatory cells for the treatment of disease. Ann. Biomed. Eng. 2014, 43, 593–602. [Google Scholar] [CrossRef]
- Zouali, M. The emerging roles of B cells as partners and targets in periodontitis. Autoimmunity 2016, 50, 61–70. [Google Scholar] [CrossRef]
- Astashina, N.; Sergeeva, E.S.; IG; Lukanin, A.N.; Kazakov, S.V. Evaluating the effectiveness of the new mouthguard design in athletes involved in power sports. Stomatologiia 2016, 95, 40–43. [Google Scholar] [CrossRef]
- Dutzan, N.; Konkel, J.E.; Greenwell-Wild, T.; Moutsopoulos, N.M. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016, 9, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.D.; Dittel, B.N.; Hardardottir, F.; Janeway, C.A. Experimental Autoimmune Encephalomyelitis Induction in Genetically B Cell–deficient Mice. J. Exp. Med. 1996, 184, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Mizoguchi, E.; Smith, R.N.; Preffer, F.I.; Bhan, A.K. Suppressive Role of B Cells in Chronic Colitis of T Cell Receptor α Mutant Mice. J. Exp. Med. 1997, 186, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune Regulatory Function of B Cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Fillatreau, S. Natural regulatory plasma cells. Curr. Opin. Immunol. 2018, 55, 62–66. [Google Scholar] [CrossRef]
- Baba, Y.; Matsumoto, M.; Kurosaki, T. Signals controlling the development and activity of regulatory B-lineage cells. Int. Immunol. 2015, 27, 487–493. [Google Scholar] [CrossRef]
- Shen, P.; Fillatreau, S. Antibody-independent functions of B cells: A focus on cytokines. Nat. Rev. Immunol. 2015, 15, 441–451. [Google Scholar] [CrossRef]
- Ray, A.; Dittel, B.N. Mechanisms of Regulatory B cell Function in Autoimmune and Inflammatory Diseases beyond IL-10. J. Clin. Med. 2017, 6, 12. [Google Scholar] [CrossRef]
- Braza, F.; Chesné, J.; Castagnet, S.; Magnan, A.; Brouard, S. Regulatory functions of B cells in allergic diseases. Allergy 2014, 69, 1454–1463. [Google Scholar] [CrossRef]
- Zhang, Y.; Gallastegui, N.; Rosenblatt, J.D. Regulatory B cells in anti-tumor immunity. Int. Immunol. 2015, 27, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Wortel, C.; Heidt, S. Regulatory B cells: Phenotype, function and role in transplantation. Transpl. Immunol. 2017, 41, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Fillatreau, S. Suppressive functions of B cells in infectious diseases: Fig. 1. Int. Immunol. 2015, 27, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Gray, D.; Mushtaq, N.; Londei, M. Prevention of Arthritis by Interleukin 10–producing B Cells. J. Exp. Med. 2003, 197, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Bouaziz, J.-D.; Matsushita, T.; Tsubata, T.; Tedder, T.F. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J. Immunol. 2009, 182, 7459–7472. [Google Scholar] [CrossRef]
- Lin, M.; Lin, J.; Wang, Y.; Bonheur, N.; Kawai, T.; Wang, Z.; Han, X. Lipopolysaccharide Attenuates CD40 Ligand-Induced Regulatory B10 Cell Expansion and IL-10 Production in Mouse Splenocytes. Open J. Immunol. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Lykken, J.M.; Candando, K.M.; Tedder, T.F. Regulatory B10 cell development and function. Int. Immunol. 2015, 27, 471–477. [Google Scholar] [CrossRef]
- Tedder, T.F. B10 Cells: A Functionally Defined Regulatory B Cell Subset. J. Immunol. 2015, 194, 1395–1401. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B Cells Maintain Regulatory T Cells While Limiting TH1 and TH17 Differentiation. Sci. Transl. Med. 2013, 5, 173ra23. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, Y.; Yu, P.; Lin, M.; Huang, G.; Kawai, T.; Taubman, M.; Wang, Z.; Xiaozhe, H. Toll-like receptor agonists Porphyromonas gingivalis LPS and CpG differentially regulate IL-10 competency and frequencies of mouse B10 cells. J. Appl. Oral Sci. 2017, 25, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Kalampokis, I.; Yoshizaki, A.; Tedder, T.F. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res. 2013, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Brown, S.; Ryan, G.; Zhao, J.; Gray, D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur. J. Immunol. 2007, 37, 3040–3053. [Google Scholar] [CrossRef] [PubMed]
- Leavy, O. B cells: IL-21 promotes B10 cell population expansion. Nat. Rev. Immunol. 2012, 12, 808–809. [Google Scholar] [CrossRef]
- Yeung, M.Y.; Ding, Q.; Brooks, C.R.; Xiao, S.; Workman, C.J.; Vignali, D.A.; Ueno, T.; Padera, R.F.; Kuchroo, V.K.; Najafian, N.; et al. TIM-1 signaling is required for maintenance and induction of regulatory B cells. Arab. Archaeol. Epigr. 2015, 15, 942–953. [Google Scholar] [CrossRef]
- Poe, J.C.; Smith, S.H.; Haas, K.M.; Yanaba, K.; Tsubata, T.; Matsushita, T.; Tedder, T.F. Amplified B Lymphocyte CD40 Signaling Drives Regulatory B10 Cell Expansion in Mice. PLoS ONE 2011, 6, 22464. [Google Scholar] [CrossRef]
- Hu, Y.; Yu, P.; Yu, X.; Hu, X.; Kawai, T.; Han, X. IL-21/anti-Tim1/CD40 ligand promotes B10 activity in vitro and alleviates bone loss in experimental periodontitis in vivo. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2149–2157. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Miyagaki, T.; DiLillo, D.J.; Matsushita, T.; Horikawa, M.; Kountikov, E.I.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Tedder, T.F. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012, 491, 264–268. [Google Scholar] [CrossRef]
- Ma, J.; Usui, Y.; Takeda, K.; Harada, N.; Yagita, H.; Okumura, K.; Akiba, H. TIM-1 signaling in B cells regulates antibody production. Biochem. Biophys. Res. Commun. 2011, 406, 223–228. [Google Scholar] [CrossRef]
- Xiao, S.; Brooks, C.R.; Sobel, R.A.; Kuchroo, V.K. Tim-1 Is Essential for Induction and Maintenance of IL-10 in Regulatory B Cells and Their Regulation of Tissue Inflammation. J. Immunol. 2015, 194, 1602–1608. [Google Scholar] [CrossRef]
- Ding, Q.; Yeung, M.; Camirand, G.; Zeng, Q.; Akiba, H.; Yagita, H.; Chalasani, G.; Sayegh, M.H.; Najafian, N.; Rothstein, D.M. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J. Clin. Investig. 2011, 121, 3645–3656. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.O.; Haynes, L.; Eaton, S.M.; Swain, S.L.; Randall, T.D. Faculty of 1000 evaluation for The biological outcome of CD40 signaling is dependent on the duration of CD40 ligand expression: Reciprocal regulation by interleukin (IL)-4 and IL-12. J. Exp. Med. 2002, 196, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; McIlraith, M.J.; Grammer, A.C.; Miura, Y.; Attrep, J.F.; Shimaoka, Y.; Lipsky, P.E. Bidirectional regulation of human B cell responses by CD40-CD40 ligand interactions. J. Immunol. 1997, 158, 4620–4633. [Google Scholar]
- Wang, Y.; Yu, X.; Lin, J.; Hu, Y.; Zhao, Q.; Kawai, T.; Taubman, M.A.; Han, X. B10 Cells Alleviate Periodontal Bone Loss in Experimental Periodontitis. Infect. Immun. 2017, 85, e00335-17. [Google Scholar] [CrossRef]
- Yu, P.; Hu, Y.; Liu, Z.; Kawai, T.; Taubman, M.A.; Li, W.; Han, X. Local Induction of B Cell Interleukin-10 Competency Alleviates Inflammation and Bone Loss in Ligature-Induced Experimental Periodontitis in Mice. Infect. Immun. 2016, 85, e00645-16. [Google Scholar] [CrossRef]
- Shi, T.; Jin, Y.; Miao, Y.; Wang, Y.; Zhou, Y.; Lin, X. IL-10 secreting B cells regulate periodontal immune response during periodontitis. Odontology 2019, 1–8. [Google Scholar] [CrossRef]
- Zhang, J.; Lapato, A.; Bodhankar, S.; Vandenbark, A.A.; Offner, H. Treatment with IL-10 producing B cells in combination with E2 ameliorates EAE severity and decreases CNS inflammation in B cell-deficient mice. Metab. Brain Dis. 2015, 30, 1117–1127. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. Human regulatory B cells in health and disease: Therapeutic potential. J. Clin. Investig. 2017, 127, 772–779. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. The expanding family of regulatory B cells. Int. Immunol. 2015, 27, 479–486. [Google Scholar] [CrossRef]
- Hong, M.; Liao, Y.; Liang, J.; Chen, X.; Li, S.; Liu, W.; Gao, C.; Zhong, Z.; Kong, D.; Deng, J.; et al. Immunomodulation of human CD19+CD25high regulatory B cells via Th17/Foxp3 regulatory T cells and Th1/Th2 cytokines. Hum. Immunol. 2019, 80, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Mohib, K.; Cherukuri, A.; Zhou, Y.; Ding, Q.; Watkins, S.C.; Rothstein, D.M. Antigen-dependent interactions between regulatory B cells and T cells at the T:B border inhibit subsequent T cell interactions with DCs. Am. J. Transplant. 2020, 20, 52–63. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Han, X. Toll-Like Receptor Signaling and Immune Regulatory Lymphocytes in Periodontal Disease. Int. J. Mol. Sci. 2020, 21, 3329. https://doi.org/10.3390/ijms21093329
Gu Y, Han X. Toll-Like Receptor Signaling and Immune Regulatory Lymphocytes in Periodontal Disease. International Journal of Molecular Sciences. 2020; 21(9):3329. https://doi.org/10.3390/ijms21093329
Chicago/Turabian StyleGu, Yingzhi, and Xiaozhe Han. 2020. "Toll-Like Receptor Signaling and Immune Regulatory Lymphocytes in Periodontal Disease" International Journal of Molecular Sciences 21, no. 9: 3329. https://doi.org/10.3390/ijms21093329
APA StyleGu, Y., & Han, X. (2020). Toll-Like Receptor Signaling and Immune Regulatory Lymphocytes in Periodontal Disease. International Journal of Molecular Sciences, 21(9), 3329. https://doi.org/10.3390/ijms21093329