The Potential Functional Roles of NME1 Histidine Kinase Activity in Neuroblastoma Pathogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
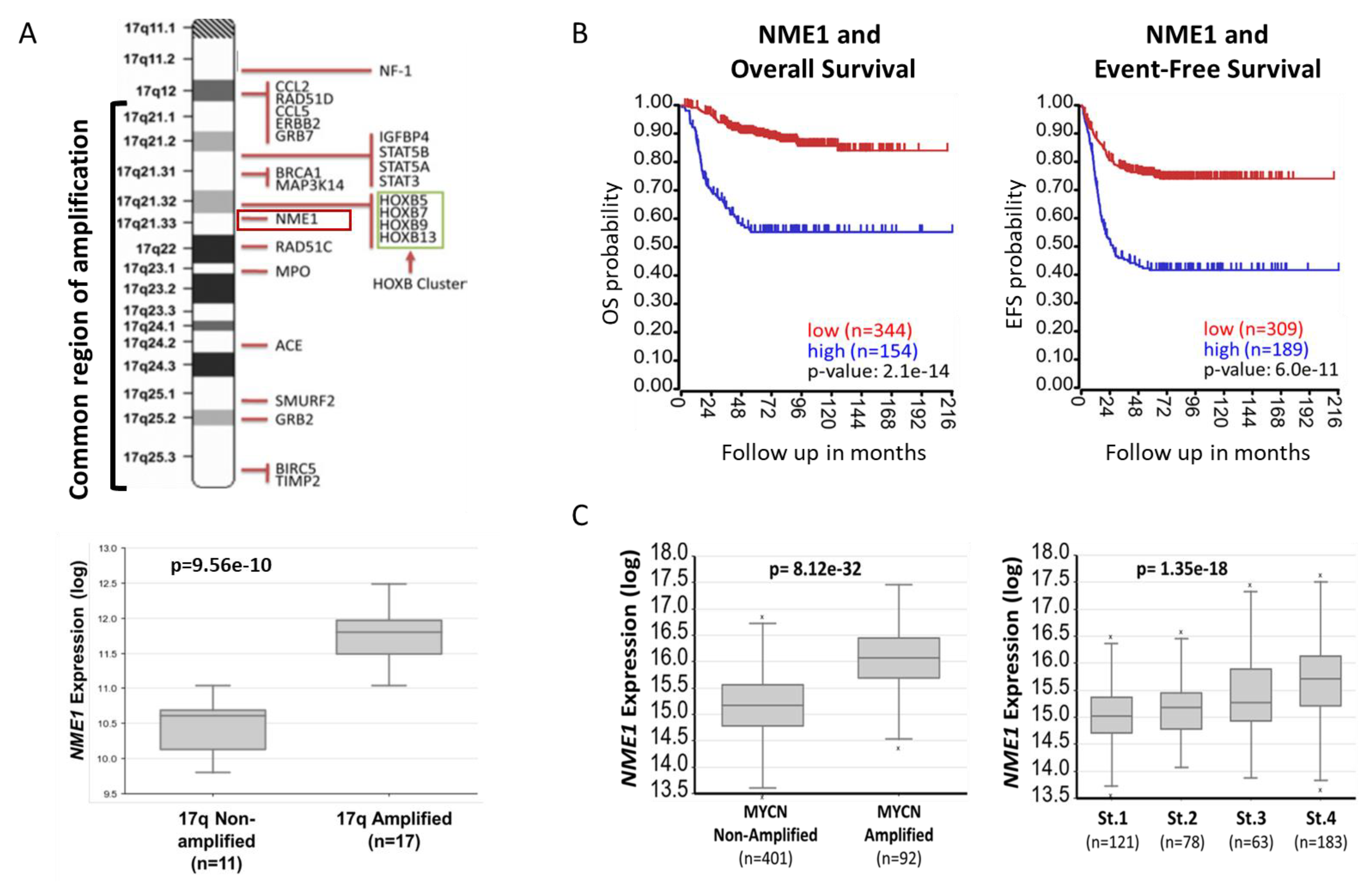
2.1. NME1 Expression Is Associated with Neuroblastoma Patient Outcomes and Prognostic Features
2.2. Histidine Phosphorylation in Neuroblastoma Cells and Tumors
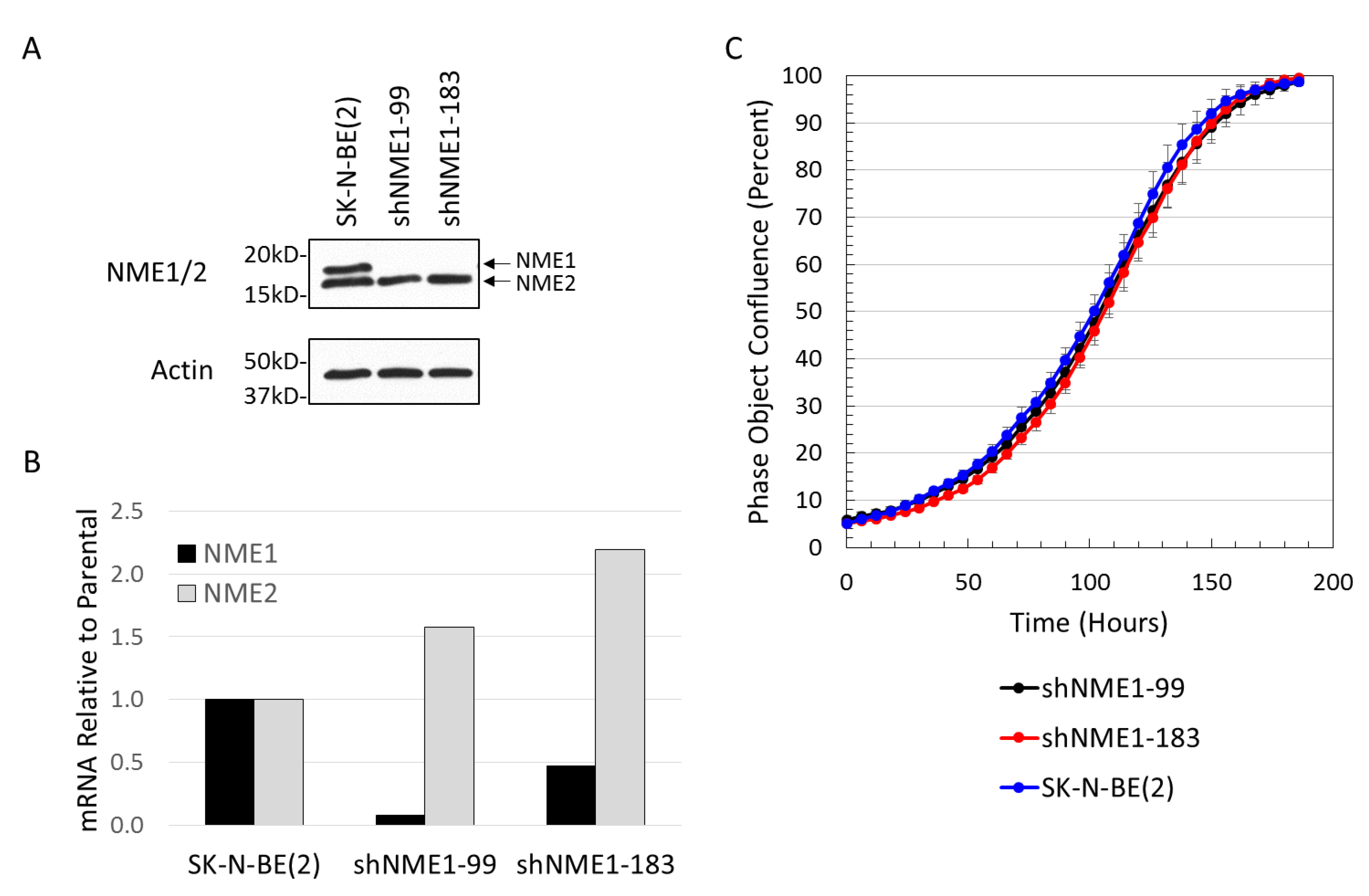
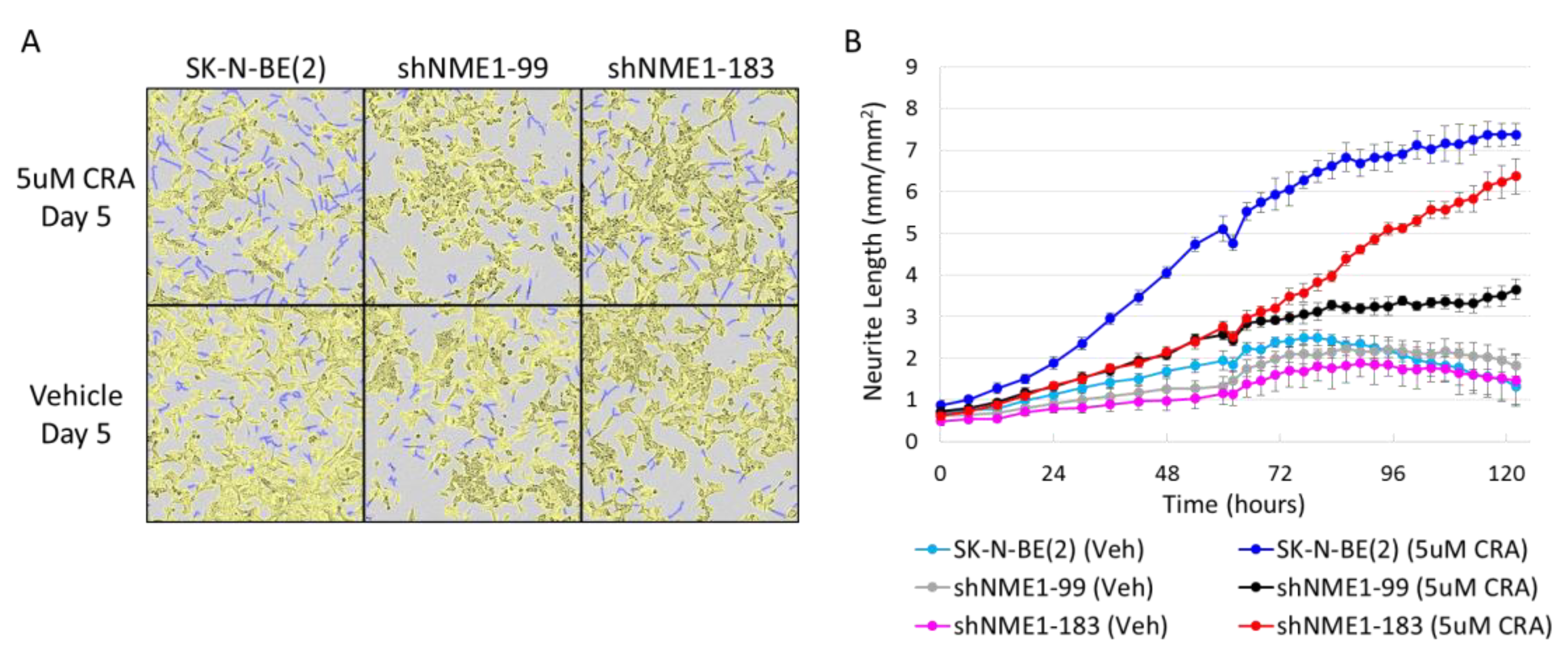
2.3. NME1 Depletion Impacts Migration and Differentiation but not Proliferation in Neuroblastoma Cells
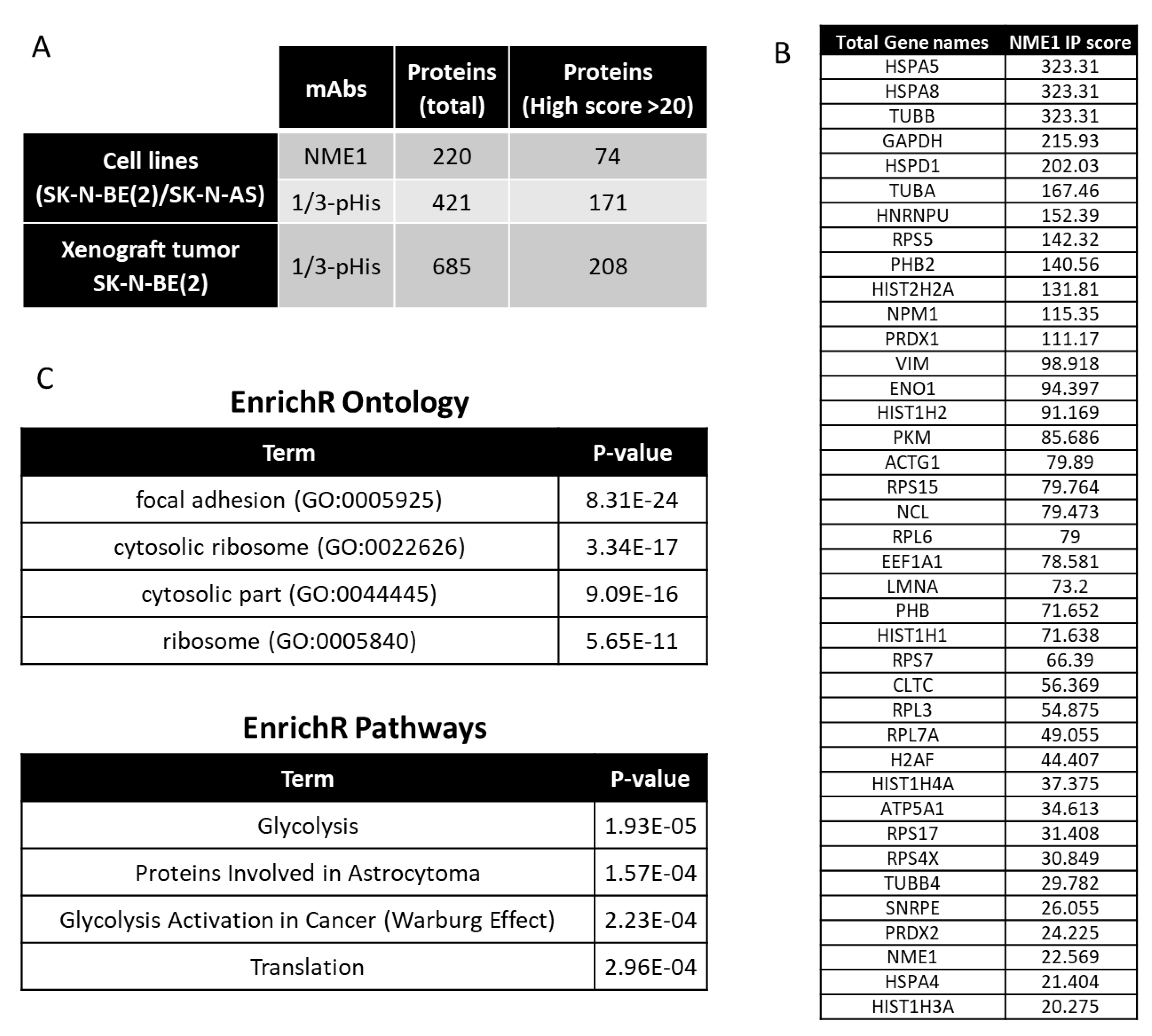
2.4. Interacting Partners of NME1 and Possible Signaling in Neuroblastoma
3. Discussion
4. Materials and Methods
4.1. Immunoblotting
4.2. Cell Culture and Stable Cell Line Generation
4.3. Quantitative PCR
4.4. Cell Proliferation
4.5. Scratch Wound Cell Migration
4.6. Cell Differentiation
4.7. Immunofluorescence Staining
4.8. Protein Immunoprecipitation from Neuroblastoma Cell Lines for MS/MS
4.9. Phosphopeptide Immunoaffinity Purification from Xenograft Tumors for MS/MS
4.10. LC-MS/MS
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| ATCC | American Type Culture Collection |
| CID | Collision Induced Dissociation |
| CRA | 13-cis-Retinoic Acid |
| EDTA | Ethylenediaminetetraacetic acid |
| EFS | Event-Free Survival |
| HCl | Hydrochloric acid |
| HPLC | High-Performance Liquid Chromatography |
| LC | Liquid Chromatography |
| LHPP | Lysine-Histidine-Pyrophosphate Phosphatase |
| MS | Mass Spectrometry |
| NDPK | Nucleoside Diphosphate Kinase |
| NH4HCO3 | Ammonium Bicarbonate |
| OCT | Optimal Cutting Temperature |
| OS | Overall Survival |
| PVDF | Polyvinylidene Difluoride |
| Q-PCR | Quantitative Polymerase Chain Reaction |
| SC | Subclone |
| SDS | Sodium Dodecyl Sulfate |
| SEQC | Cell Sequencing |
| TEA | Triethanolamine |
| TLCK | Tosyl-L-Lysyl-Chloromethane Hydrochloride |
References
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef]
- Bown, N.; Lastowska, M.; Cotterill, S.; O’Neill, S.; Ellershaw, C.; Roberts, P.; Lewis, I.; Pearson, A.D.; U.K. Cancer Cytogenetics Group and the U.K. Children’s Cancer Study Group. 17q gain in neuroblastoma predicts adverse clinical outcome. Med. Pediatr. Oncol. 2001, 36, 14–19. [Google Scholar] [CrossRef]
- Irwin, M.S.; Park, J.R. Neuroblastoma: Paradigm for precision medicine. Pediatr. Clin. N. Am. 2015, 62, 225–256. [Google Scholar] [CrossRef]
- Meddeb, M.; Danglot, G.; Chudoba, I.; Vénuat, A.M.; Bénard, J.; Avet-Loiseau, H.; Vasseur, B.; Le Paslier, D.; Terrier-Lacombe, M.J.; Hartmann, O.; et al. Additional copies of a 25 Mb chromosomal region originating from 17q23.1-17qter are present in 90% of high-grade neuroblastomas. Genes Chromosomes Cancer 1996, 17, 156–165. [Google Scholar] [CrossRef]
- Bown, N.; Cotterill, S.; Lastowska, M.; O’Neill, S.; Pearson, A.D.; Plantaz, D.; Meddeb, M.; Danglot, G.; Brinkschmidt, C.; Christiansen, H.; et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999, 340, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Plantaz, D.; Mohapatra, G.; Matthay, K.K.; Pellarin, M.; Seeger, R.C.; Feuerstein, B.G. Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization. Am. J. Pathol. 1997, 150, 81–89. [Google Scholar] [PubMed]
- Bilitou, A.; Watson, J.; Gartner, A.; Ohnuma, S. The NM23 family in development. Mol. Cell Biochem. 2009, 329, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, A.; Liu, W.; Wang, C.; Wang, M.; Zhang, L.; Wang, D.; Dong, J.; Li, M. NME2 reduces proliferation, migration and invasion of gastric cancer cells to limit metastasis. PLoS ONE 2015, 10, e0115968. [Google Scholar] [CrossRef] [PubMed]
- Owlanj, H.; Yang, H.J.; Feng, Z.W. Nucleoside diphosphate kinase Nm23-M1 involves in oligodendroglial versus neuronal cell fate decision in vitro. Differentiation 2012, 84, 281–293. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Steeg, P.S. Nm23/nucleoside diphosphate kinase in human cancers. J. Bioenerg. Biomembr. 2000, 32, 301–308. [Google Scholar] [CrossRef]
- Katakura, H.; Tanaka, F.; Oyanagi, H.; Miyahara, R.; Yanagihara, K.; Otake, Y.; Wada, H. Clinical significance of nm23 expression in resected pathologic-stage I, non-small cell lung cancer. Ann. Thorac. Surg. 2002, 73, 1060–1064. [Google Scholar] [CrossRef]
- Lombardi, D.; Lacombe, M.L.; Paggi, M.G. Nm23: Unraveling its biological function in cell differentiation. J. Cell Physiol. 2000, 182, 144–149. [Google Scholar] [CrossRef]
- Mao, H.; Liu, H.; Fu, X.; Fang, Z.; Abrams, J.; Worsham, M.J. Loss of nm23 expression predicts distal metastases and poorer survival for breast cancer. Int. J. Oncol. 2001, 18, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-S.; Chow, K.-C.; Lien, Y.-C.; Kuo, K.-T.; Li, W.-Y. Prognostic significance of nm23-H1 expression in esophageal squamous cell carcinoma. Eur. J. Cardiothorac. Surg. 2004, 26, 419–424. [Google Scholar] [CrossRef]
- Guan-Zhen, Y.; Ying, C.; Can-Rong, N.; Guo-Dong, W.; Jian-Xin, Q.; Jie-Jun, W. Reduced protein expression of metastasis-related genes (nm23, KISS1, KAI1 and p53) in lymph node and liver metastases of gastric cancer. Int. J. Exp. Pathol. 2007, 88, 175–183. [Google Scholar] [CrossRef]
- Niu, Y.; Fu, X.; Lv, A.; Fan, Y.; Wang, Y. Potential markers predicting distant metastasis in axillary node-negative breast carcinoma. Int. J. Cancer 2002, 98, 754–760. [Google Scholar] [CrossRef][Green Version]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef]
- Terasaki-Fukuzawa, Y.; Kijima, H.; Suto, A.; Takeshita, T.; Iezumi, K.; Sato, S.; Yoshida, H.; Sato, T.; Shimbori, M.; Shiina, Y. Decreased nm23 expression, but not Ki-67 labeling index, is significantly correlated with lymph node metastasis of breast invasive ductal carcinoma. Int. J. Mol. Med. 2002, 9, 25–29. [Google Scholar] [CrossRef]
- Yoshida, H.; Kijima, H.; Terasaki, Y.; Suto, A.; Takeshita, T.; Omiya, H.; Shimojima, K.; Shimbori, M.; Sato, T.; Sato, S.; et al. Heterogeneous expression of nm23 gene product as a predictor of lymph nodal status in human breast cancer. Int. J. Oncol. 1998, 13, 1141–1146. [Google Scholar] [CrossRef]
- McCorkle, J.R.; Leonard, M.K.; Kraner, S.D.; Blalock, E.M.; Ma, D.; Zimmer, S.G.; Kaetzel, D.M. The metastasis suppressor NME1 regulates expression of genes linked to metastasis and patient outcome in melanoma and breast carcinoma. Cancer Genom. Proteom. 2014, 11, 175–194. [Google Scholar]
- Boissan, M.; Wendum, D.; Arnaud-Dabernat, S.; Munier, A.; Debray, M.; Lascu, I.; Daniel, J.-Y.; Lacombe, M.-L. Increased lung metastasis in transgenic NM23-Null/SV40 mice with hepatocellular carcinoma. J. Natl. Cancer Inst. 2005, 97, 836–845. [Google Scholar] [CrossRef]
- Boissan, M.; De Wever, O.; Lizarraga, F.; Wendum, D.; Poincloux, R.; Chignard, N.; Desbois-Mouthon, C.; Dufour, S.; Nawrocki-Raby, B.; Birembaut, P.; et al. Implication of metastasis suppressor NM23-H1 in maintaining adherens junctions and limiting the invasive potential of human cancer cells. Cancer Res. 2010, 70, 7710–7722. [Google Scholar] [CrossRef]
- De Preter, K.; Vermeulen, J.; Brors, B.; Delattre, O.; Eggert, A.; Fischer, M.; Janoueix-Lerosey, I.; Lavarino, C.; Maris, J.M.; Mora, J.; et al. Accurate outcome prediction in neuroblastoma across independent data sets using a multigene signature. Clin. Cancer Res. 2010, 16, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Ohira, M.; Oba, S.; Nakamura, Y.; Isogai, E.; Kaneko, S.; Nakagawa, A.; Hirata, T.; Kubo, H.; Goto, T.; Yamada, S.; et al. Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell 2005, 7, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.S.; Greer, B.T.; Westermann, F.; Steinberg, S.M.; Son, C.-G.; Chen, Q.-R.; Whiteford, C.C.; Bilke, S.; Krasnoselsky, A.L.; Cenacchi, N.; et al. Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res. 2004, 64, 6883–6891. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Mayol, G.; Ríos, J.; Domenech, G.; Cheung, N.-K.V.; Oberthuer, A.; Fischer, M.; Maris, J.M.; Brodeur, G.M.; Hero, B.; et al. A three-gene expression signature model for risk stratification of patients with neuroblastoma. Clin. Cancer Res. 2012, 18, 2012–2023. [Google Scholar] [CrossRef]
- Lecroisey, A.; Lascu, I.; Bominaar, A.; Véron, M.; Delepierre, M. Phosphorylation mechanism of nucleoside diphosphate kinase: 31P-nuclear magnetic resonance studies. Biochemistry 1995, 34, 12445–12450. [Google Scholar] [CrossRef]
- Adam, K.; Hunter, T. Histidine kinases and the missing phosphoproteome from prokaryotes to eukaryotes. Lab. Invest. 2017, 98, 233–247. [Google Scholar] [CrossRef]
- Achyutuni, S.; Nadhan, R.; Sengodan, S.; Srinivas, P. The prodigious network of chromosome 17 miRNAs regulating cancer genes that influence the hallmarks of cancer. Semin. Oncol. 2017, 44, 254–264. [Google Scholar] [CrossRef]
- Łastowska, M.; Cotterill, S.; Pearson, A.D.J.; Roberts, P.; McGuckin, A.; Lewis, I.; Bown, N. Gain of chromosome arm 17q predicts unfavourable outcome in neuroblastoma patients. Eur. J. Cancer 1997, 33, 1627–1633. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16. [Google Scholar] [CrossRef]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.K.Y.; Betz, C.; et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature 2018, 555, 678–682. [Google Scholar] [CrossRef]
- Fuhs, S.R.; Meisenhelder, J.; Aslanian, A.; Ma, L.; Zagorska, A.; Stankova, M.; Binnie, A.; Al-Obeidi, F.; Mauger, J.; Lemke, G.; et al. Monoclonal 1- and 3-phosphohistidine antibodies: New tools to study histidine phosphorylation. Cell 2015, 162, 198–210. [Google Scholar] [CrossRef]
- Adam, K.; Fuhs, S.; Meisenhelder, J.; Aslanian, A.; Diedrich, J.; Moresco, J.; Clair, J.L.; Yates, J.R.; Hunter, T. A non-acidic method using hydroxyapatite and phosphohistidine monoclonal antibodies allows enrichment of phosphopeptides containing non-conventional phosphorylations for mass spectrometry analysis. BioRxiv 2019, 691352. [Google Scholar]
- Engel, M.; Seifert, M.; Theisinger, B.; Seyfert, U.; Welter, C. Glyceraldehyde-3-phosphate dehydrogenase and Nm23-H1/nucleoside diphosphate kinase A. Two old enzymes combine for the novel Nm23 protein phosphotransferase function. J. Biol. Chem. 1998, 273, 20058–20065. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Lu, Q.; Park, H.; Egger, L.A.; Inouye, M. Nucleoside-diphosphate kinase-mediated signal transduction via histidyl-aspartyl phosphorelay systems in Escherichia coli. J. Biol. Chem. 1996, 271, 32886–32893. [Google Scholar] [CrossRef]
- Mehta, A.; Orchard, S. Nucleoside diphosphate kinase (NDPK, NM23, AWD): Recent regulatory advances in endocytosis, metastasis, psoriasis, insulin release, fetal erythroid lineage and heart failure; translational medicine exemplified. Mol. Cell Biochem. 2009, 329, 3–15. [Google Scholar] [CrossRef][Green Version]
- Nallamothu, G.; Dammai, V.; Hsu, T. Developmental function of Nm23/awd: A mediator of endocytosis. Mol. Cell Biochem. 2009, 329, 35–44. [Google Scholar] [CrossRef][Green Version]
- Rochdi, M.D.; Laroche, G.; Dupré, E.; Giguère, P.; Lebel, A.; Watier, V.; Hamelin, E.; Lépine, M.-C.; Dupuis, G.; Parent, J.-L. Nm23-H2 interacts with a G protein-coupled receptor to regulate its endocytosis through an Rac1-dependent mechanism. J. Biol. Chem. 2004, 279, 18981–18989. [Google Scholar] [CrossRef]
- Almgren, M.A.E.; Henriksson, K.C.E.; Fujimoto, J.; Chang, C.L. Nucleoside diphosphate kinase A/nm23-H1 promotes metastasis of NB69-derived human neuroblastoma. Mol. Cancer Res. 2004, 2, 387–394. [Google Scholar]
- Tan, C.-Y.; Chang, C.L. NDPKA is not just a metastasis suppressor—Be aware of its metastasis-promoting role in neuroblastoma. Lab. Invest. 2017, 98, 219–227. [Google Scholar] [CrossRef]
- Leone, A.; Seeger, R.C.; Hong, C.M.; Hu, Y.Y.; Arboleda, M.J.; Brodeur, G.M.; Stram, D.; Slamon, D.J.; Steeg, P.S. Evidence for nm23 RNA overexpression, DNA amplification and mutation in aggressive childhood neuroblastomas. Oncogene 1993, 8, 855–865. [Google Scholar]
- Takeda, O.; Handa, M.; Uehara, T.; Maseki, N.; Sakashita, A.; Sakurai, M.; Kanda, N.; Arai, Y.; Kaneko, Y. An increased NM23H1 copy number may be a poor prognostic factor independent of LOH on 1p in neuroblastomas. Br. J. Cancer 1996, 74, 1620–1626. [Google Scholar] [CrossRef][Green Version]
- Caron, H. Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med. Pediatr. Oncol. 1995, 24, 215–221. [Google Scholar] [CrossRef]
- Hailat, N.; Keim, D.R.; Melhem, R.F.; Zhu, X.X.; Eckerskorn, C.; Brodeur, G.M.; Reynolds, C.P.; Seeger, R.C.; Lottspeich, F.; Strahler, J.R. High levels of p19/nm23 protein in neuroblastoma are associated with advanced stage disease and with N-myc gene amplification. J. Clin. Invest. 1991, 88, 341–345. [Google Scholar] [CrossRef]
- Ohira, M.; Morohashi, A.; Inuzuka, H.; Shishikura, T.; Kawamoto, T.; Kageyama, H.; Nakamura, Y.; Isogai, E.; Takayasu, H.; Sakiyama, S.; et al. Expression profiling and characterization of 4200 genes cloned from primary neuroblastomas: Identification of 305 genes differentially expressed between favorable and unfavorable subsets. Oncogene 2003, 22, 5525–5536. [Google Scholar] [CrossRef]
- Godfried, M.B.; Veenstra, M.; v Sluis, P.; Boon, K.; v Asperen, R.; Hermus, M.C.; v Schaik, B.D.C.; Voûte, T.P.A.; Schwab, M.; Versteeg, R.; et al. The N-myc and c-myc downstream pathways include the chromosome 17q genes nm23-H1 and nm23-H2. Oncogene 2002, 21, 2097–2101. [Google Scholar] [CrossRef]
- Chang, C.L.; Zhu, X.X.; Thoraval, D.H.; Ungar, D.; Rawwas, J.; Hora, N.; Strahler, J.R.; Hanash, S.M.; Radany, E. Nm23-H1 mutation in neuroblastoma. Nature 1994, 370, 335–336. [Google Scholar] [CrossRef]
- Wagner, P.D.; Steeg, P.S.; Vu, N.D. Two-component kinase-like activity of nm23 correlates with its motility-suppressing activity. Proc. Natl. Acad. Sci. USA 1997, 94, 9000–9005. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Cuello, F.; Schulze, R.A.; Heemeyer, F.; Meyer, H.E.; Lutz, S.; Jakobs, K.H.; Niroomand, F.; Wieland, T. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Complex formation of NDPK B with Gbeta gamma dimers and phosphorylation of His-266 IN Gbeta. J. Biol. Chem. 2003, 278, 7220–7226. [Google Scholar] [CrossRef]
- Giraud, M.-F.; Georgescauld, F.; Lascu, I.; Dautant, A. Crystal structures of S120G mutant and wild type of human nucleoside diphosphate kinase A in complex with ADP. J. Bioenerg. Biomembr. 2006, 38, 261–264. [Google Scholar] [CrossRef]
- Morera, S.; Lascu, I.; Dumas, C.; LeBras, G.; Briozzo, P.; Veron, M.; Janin, J. Adenosine 5′-diphosphate binding and the active site of nucleoside diphosphate kinase. Biochemistry 1994, 33, 459–467. [Google Scholar] [CrossRef]
- Engel, M.; Véron, M.; Theisinger, B.; Lacombe, M.L.; Seib, T.; Dooley, S.; Welter, C. A novel serine/threonine-specific protein phosphotransferase activity of Nm23/nucleoside-diphosphate kinase. Eur. J. Biochem. 1995, 234, 200–207. [Google Scholar] [CrossRef]
- Ma, D.; McCorkle, J.R.; Kaetzel, D.M. The metastasis suppressor NM23-H1 possesses 3′-5′ exonuclease activity. J. Biol. Chem. 2004, 279, 18073–18084. [Google Scholar] [CrossRef]
- Wagner, P.D.; Vu, N.D. Phosphorylation of geranyl and farnesyl pyrophosphates by Nm23 proteins/nucleoside diphosphate kinases. J. Biol. Chem. 2000, 275, 35570–35576. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Singh, P.; Lee, D.-H.; Qiu, J.; Cai, S.; O’Connor, T.R.; Chen, Y.; Shen, B.; Pfeifer, G.P. Characterization of the 3′ --> 5′ exonuclease activity found in human nucleoside diphosphate kinase 1 (NDK1) and several of its homologues. Biochemistry 2005, 44, 15774–15786. [Google Scholar] [CrossRef]
- Cai, X.; Srivastava, S.; Surindran, S.; Li, Z.; Skolnik, E.Y. Regulation of the epithelial Ca2+ channel TRPV5 by reversible histidine phosphorylation mediated by NDPK-B and PHPT1. Mol. Biol. Cell 2014, 25, 1244–1250. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Morrison, D.K.; Salerno, M.; Palmieri, D.; Ouatas, T.; Mair, M.; Patrick, J.; Steeg, P.S. Nm23-H1 metastasis suppressor phosphorylation of kinase suppressor of Ras via a histidine protein kinase pathway. J. Biol. Chem. 2002, 277, 32389–32399. [Google Scholar] [CrossRef]
- Muimo, R.; Hornickova, Z.; Riemen, C.E.; Gerke, V.; Matthews, H.; Mehta, A. Histidine phosphorylation of annexin I in airway epithelia. J. Biol. Chem. 2000, 275, 36632–36636. [Google Scholar] [CrossRef]
- Srivastava, S.; Li, Z.; Ko, K.; Choudhury, P.; Albaqumi, M.; Johnson, A.K.; Yan, Y.; Backer, J.M.; Unutmaz, D.; Coetzee, W.A.; et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell 2006, 24, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.D.; Vu, N.D. Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J. Biol. Chem. 1995, 270, 21758–21764. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.D.; Vu, N.D. Histidine to aspartate phosphotransferase activity of nm23 proteins: Phosphorylation of aldolase C on Asp-319. Biochem. J. 2000, 346, 623–630. [Google Scholar] [CrossRef]
- Wieland, T.; Attwood, P.V. Alterations in reversible protein histidine phosphorylation as intracellular signals in cardiovascular disease. Front. Pharmacol. 2015, 6, 173. [Google Scholar] [CrossRef]
- Boissan, M.; Dabernat, S.; Peuchant, E.; Schlattner, U.; Lascu, I.; Lacombe, M.-L. The mammalian Nm23/NDPK family: From metastasis control to cilia movement. Mol. Cell Biochem. 2009, 329, 51–62. [Google Scholar] [CrossRef]
- Webb, P.A.; Perisic, O.; Mendola, C.E.; Backer, J.M.; Williams, R.L. The crystal structure of a human nucleoside diphosphate kinase, NM23-H2. J. Mol. Biol. 1995, 251, 574–587. [Google Scholar] [CrossRef]
- Boissan, M.; Lacombe, M.-L. Learning about the functions of NME/NM23: Lessons from knockout mice to silencing strategies. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 384, 421–431. [Google Scholar] [CrossRef]
- Postel, E.H.; Wohlman, I.; Zou, X.; Juan, T.; Sun, N.; D’Agostin, D.; Cuellar, M.; Choi, T.; Notterman, D.A.; Perle, K.M.D.L. Targeted deletion of Nm23/nucleoside diphosphate kinase A and B reveals their requirement for definitive erythropoiesis in the mouse embryo. Dev. Dyn. 2009, 238, 775–787. [Google Scholar] [CrossRef]
- Milon, L.; Meyer, P.; Chiadmi, M.; Munier, A.; Johansson, M.; Karlsson, A.; Lascu, I.; Capeau, J.; Janin, J.; Lacombe, M.L. The human nm23-H4 gene product is a mitochondrial nucleoside diphosphate kinase. J. Biol. Chem. 2000, 275, 14264–14272. [Google Scholar] [CrossRef]
- Liu, P.; Choi, Y.-K.; Qi, R.Z. NME7 is a functional component of the γ-tubulin ring complex. Mol. Biol. Cell 2014, 25, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, M.; Pedone, E.; Diana, D.; de Antonellis, P.; Džeroski, S.; Marino, N.; Navas, L.; Di Dato, V.; Scoppettuolo, M.N.; Cimmino, F.; et al. Neuroblastoma tumorigenesis is regulated through the Nm23-H1/h-Prune C-terminal interaction. Sci. Rep. 2013, 3, 1351. [Google Scholar] [CrossRef]
- D’Angelo, A.; Garzia, L.; André, A.; Carotenuto, P.; Aglio, V.; Guardiola, O.; Arrigoni, G.; Cossu, A.; Palmieri, G.; Aravind, L.; et al. Prune cAMP phosphodiesterase binds nm23-H1 and promotes cancer metastasis. Cancer Cell 2004, 5, 137–149. [Google Scholar] [CrossRef]
- Subramanian, C.; Cotter, M.A.; Robertson, E.S. Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-H1: A molecular link to cancer metastasis. Nat. Med. 2001, 7, 350–355. [Google Scholar] [CrossRef]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G.; et al. Membrane trafficking. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef]
- Moreno, V.; Gonzalo, P.; Gómez-Escudero, J.; Pollán, Á.; Acín-Pérez, R.; Breckenridge, M.; Yáñez-Mó, M.; Barreiro, O.; Orsenigo, F.; Kadomatsu, K.; et al. An EMMPRIN-γ-catenin-Nm23 complex drives ATP production and actomyosin contractility at endothelial junctions. J. Cell Sci. 2014, 127, 3768–3781. [Google Scholar] [CrossRef]
- Adam, K.; Hunter, T. Subcellular localization of histidine phosphorylated proteins through indirect immunofluorescence. Methods Mol. Biol. 2020, 2077, 209–224. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adam, K.; Lesperance, J.; Hunter, T.; Zage, P.E. The Potential Functional Roles of NME1 Histidine Kinase Activity in Neuroblastoma Pathogenesis. Int. J. Mol. Sci. 2020, 21, 3319. https://doi.org/10.3390/ijms21093319
Adam K, Lesperance J, Hunter T, Zage PE. The Potential Functional Roles of NME1 Histidine Kinase Activity in Neuroblastoma Pathogenesis. International Journal of Molecular Sciences. 2020; 21(9):3319. https://doi.org/10.3390/ijms21093319
Chicago/Turabian StyleAdam, Kevin, Jacqueline Lesperance, Tony Hunter, and Peter E. Zage. 2020. "The Potential Functional Roles of NME1 Histidine Kinase Activity in Neuroblastoma Pathogenesis" International Journal of Molecular Sciences 21, no. 9: 3319. https://doi.org/10.3390/ijms21093319
APA StyleAdam, K., Lesperance, J., Hunter, T., & Zage, P. E. (2020). The Potential Functional Roles of NME1 Histidine Kinase Activity in Neuroblastoma Pathogenesis. International Journal of Molecular Sciences, 21(9), 3319. https://doi.org/10.3390/ijms21093319