Therapeutic Application of Drug-Conjugated HER2 Oligobody (HER2-DOligobody)

 ,
,  and
and 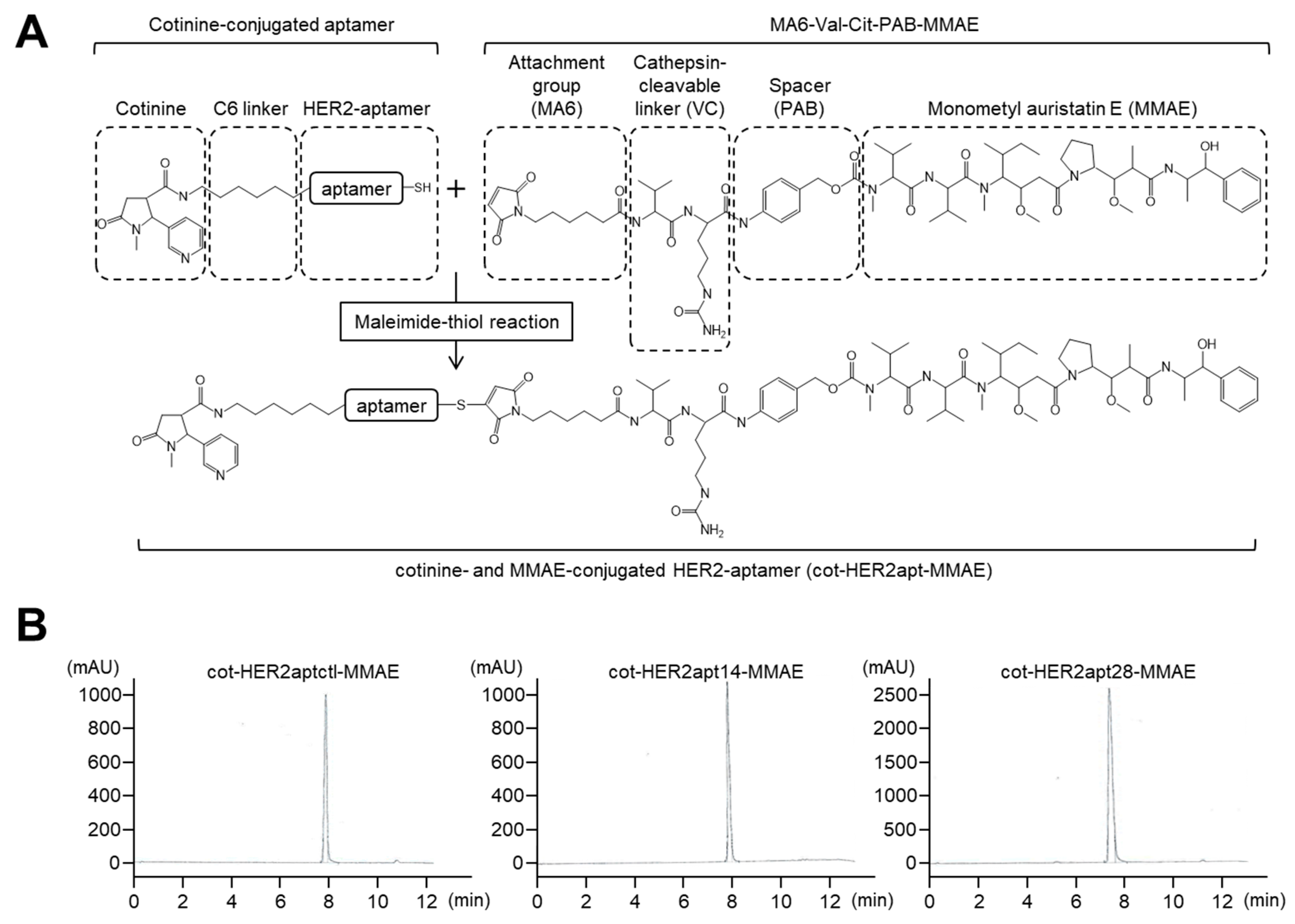{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
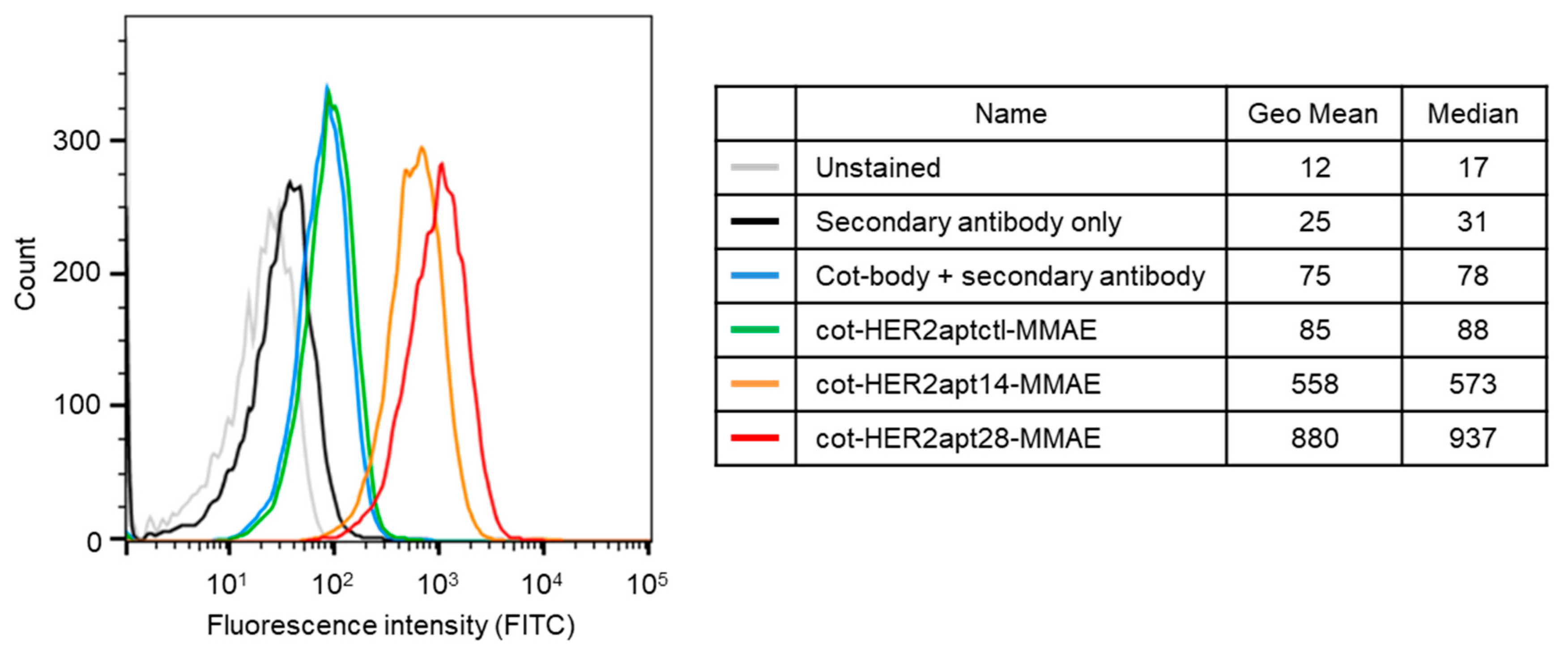
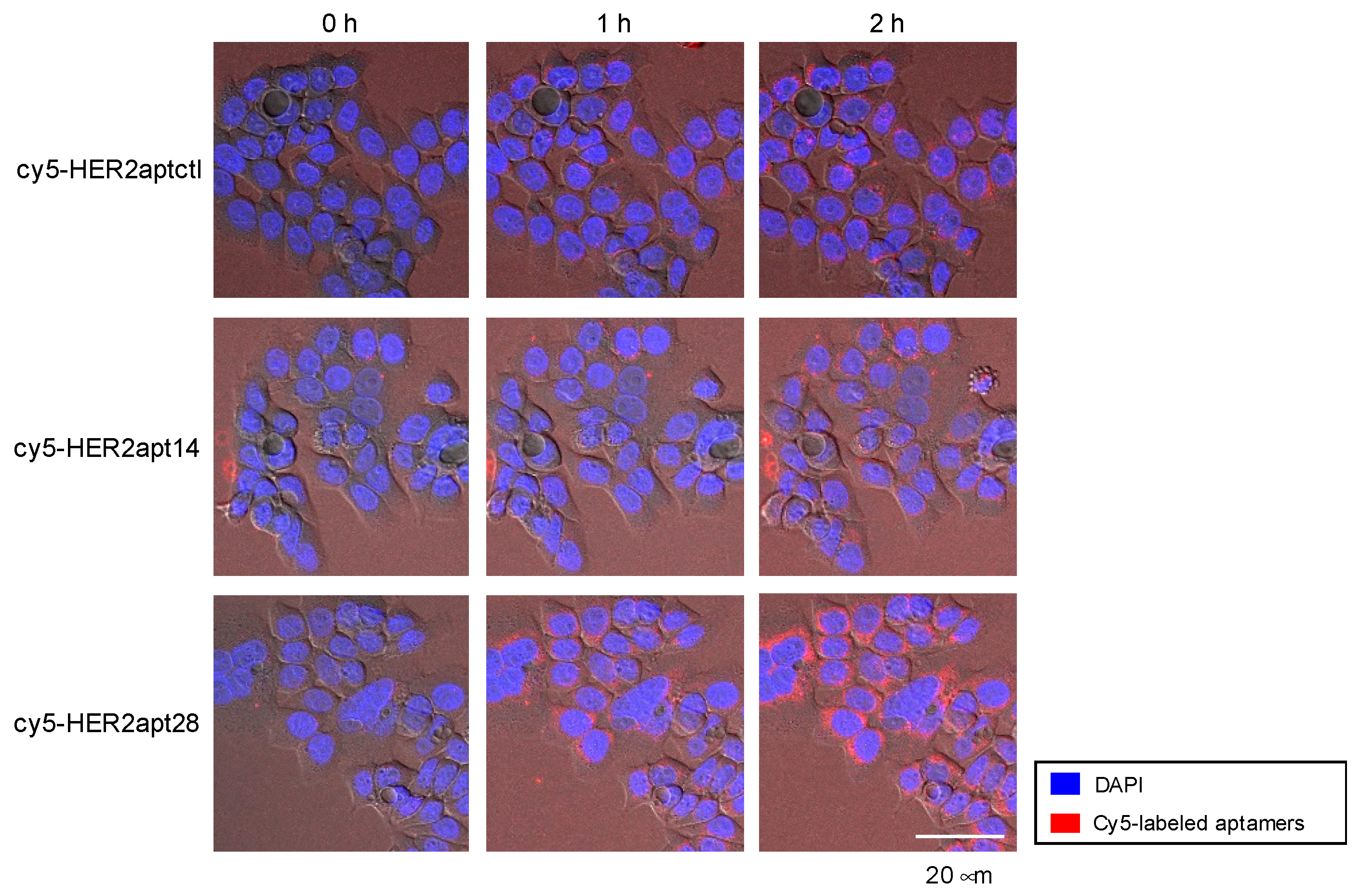
2.1. Characterization of HER2 Aptamers and Cotinine- and MMAE-Conjugated HER2-Aptamers
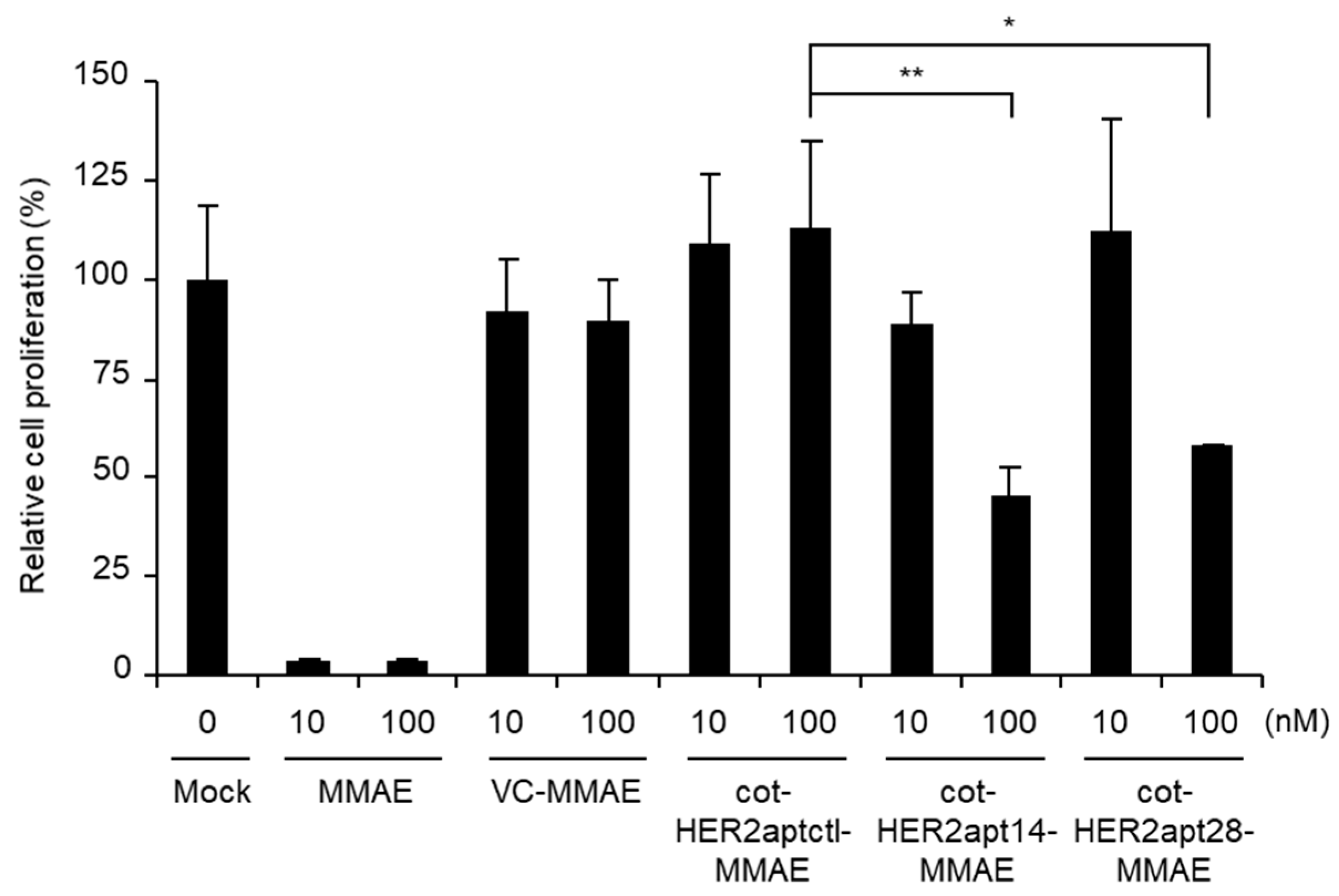
2.2. Antitumor Activity of cot-HER2apt-MMAEs In Vitro
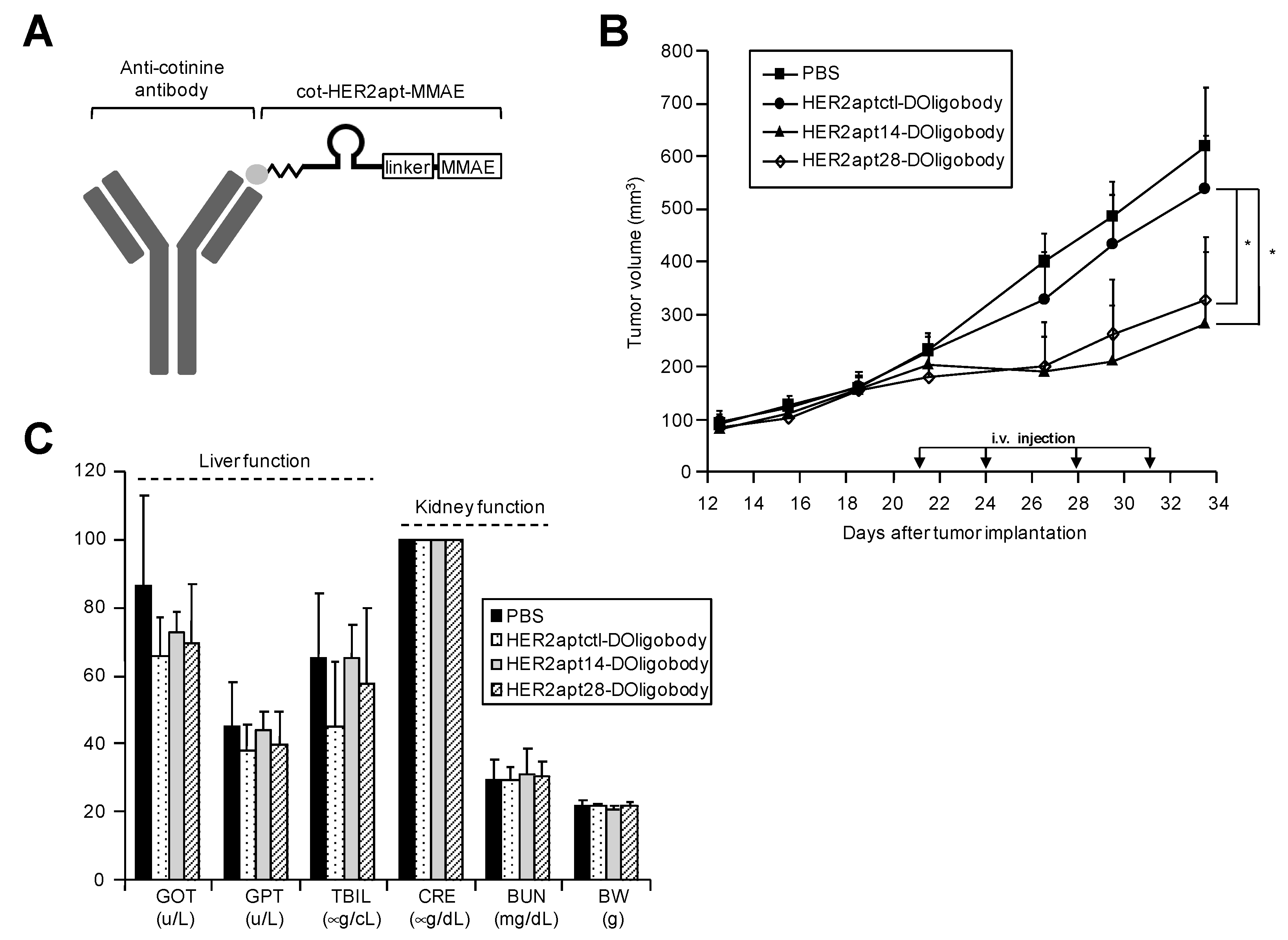
2.3. Antitumor Activity of HER2-DOligobodies In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Synthesis of cot-HER2apt-MMAEs and HER2-DOligobodies
4.3. Flow Cytometry (FACS)
4.4. Confocal Imaging
4.5. Cell Proliferation Assay
4.6. Nude Mouse Xenograft Model of Gastric Cancer
4.7. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Valge-Archer, V.E. Development trends for monoclonal antibody cancer therapeutics. Nat. Rev. Drug Discov 2007, 6, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Lambert, J.M. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr. Opin. Pharmacol. 2005, 5, 543–549. [Google Scholar] [CrossRef]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- Scott, L.J. Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs 2017, 77, 435–445. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab Emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- McGregor, B.A.; Sonpavde, G. Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem. Pharm. Bull. (Tokyo) 2019, 67, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Wang, L.; Amphlett, G.; Blattler, W.A.; Lambert, J.M.; Zhang, W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Shigdar, S.; Macdonald, J.; O’Connor, M.; Wang, T.; Xiang, D.; Al Shamaileh, H.; Qiao, L.; Wei, M.; Zhou, S.F.; Zhu, Y.; et al. Aptamers as theranostic agents: Modifications, serum stability and functionalisation. Sensors (Basel) 2013, 13, 13624–13637. [Google Scholar] [CrossRef]
- Song, K.M.; Lee, S.; Ban, C. Aptamers and their biological applications. Sensors (Basel) 2012, 12, 612–631. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.A.; Lopez-Colon, D.; Zhu, Z.; Xu, Y.; Tan, W. Applications of aptamers in cancer cell biology. Anal. Chim Acta 2008, 621, 101–108. [Google Scholar] [CrossRef]
- Watson, S.R.; Chang, Y.F.; O’Connell, D.; Weigand, L.; Ringquist, S.; Parma, D.H. Anti-L-selectin aptamers: Binding characteristics, pharmacokinetic parameters, and activity against an intravascular target in vivo. Antisense Nucleic Acid Drug Dev. 2000, 10, 63–75. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Keating, G.M. Pegaptanib: In exudative age-related macular degeneration. Drugs 2005, 65, 1571–1577, discussion 1578-1579. [Google Scholar] [CrossRef]
- Heo, K.; Min, S.W.; Sung, H.J.; Kim, H.G.; Kim, H.J.; Kim, Y.H.; Choi, B.K.; Han, S.; Chung, S.; Lee, E.S.; et al. An aptamer-antibody complex (oligobody) as a novel delivery platform for targeted cancer therapies. J. Control. Release 2016, 229, 1–9. [Google Scholar] [CrossRef]
- Pettit, G.R. The dolastatins. Fortschr. Chem. Org. Naturst. 1997, 70, 1–79. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Mahlknecht, G.; Maron, R.; Mancini, M.; Schechter, B.; Sela, M.; Yarden, Y. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc. Natl. Acad. Sci. USA 2013, 110, 8170–8175. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy: Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Factors determining antibody distribution in tumors. Trends Pharmacol. Sci. 2008, 29, 57–61. [Google Scholar] [CrossRef]
- Xiang, D.; Shigdar, S.; Qiao, G.; Wang, T.; Kouzani, A.Z.; Zhou, S.F.; Kong, L.; Li, Y.; Pu, C.; Duan, W. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: The next generation of cancer medicine. Theranostics 2015, 5, 23–42. [Google Scholar] [CrossRef]
- White, R.R.; Sullenger, B.A.; Rusconi, C.P. Developing aptamers into therapeutics. J. Clin. Invest. 2000, 106, 929–934. [Google Scholar] [CrossRef]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.S.; Cook, G.; Warren, S.; et al. Tumor targeting by an aptamer. J. Nucl Med. 2006, 47, 668–678. [Google Scholar]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Park, S.; Hwang, D.; Chung, J. Cotinine-conjugated aptamer/anti-cotinine antibody complexes as a novel affinity unit for use in biological assays. Exp. Mol. Med. 2012, 44, 554–561. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Sung, H.J.; Lee, Y.M.; Choi, S.I.; Kim, Y.-H.; Heo, K.; Kim, I.-H. Therapeutic Application of Drug-Conjugated HER2 Oligobody (HER2-DOligobody). Int. J. Mol. Sci. 2020, 21, 3286. https://doi.org/10.3390/ijms21093286
Kim HJ, Sung HJ, Lee YM, Choi SI, Kim Y-H, Heo K, Kim I-H. Therapeutic Application of Drug-Conjugated HER2 Oligobody (HER2-DOligobody). International Journal of Molecular Sciences. 2020; 21(9):3286. https://doi.org/10.3390/ijms21093286
Chicago/Turabian StyleKim, Hyun Jung, Ho Jin Sung, Yul Min Lee, Sun Il Choi, Yun-Hee Kim, Kyun Heo, and In-Hoo Kim. 2020. "Therapeutic Application of Drug-Conjugated HER2 Oligobody (HER2-DOligobody)" International Journal of Molecular Sciences 21, no. 9: 3286. https://doi.org/10.3390/ijms21093286
APA StyleKim, H. J., Sung, H. J., Lee, Y. M., Choi, S. I., Kim, Y.-H., Heo, K., & Kim, I.-H. (2020). Therapeutic Application of Drug-Conjugated HER2 Oligobody (HER2-DOligobody). International Journal of Molecular Sciences, 21(9), 3286. https://doi.org/10.3390/ijms21093286