Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells


Abstract
1. Introduction
2. Replicative Senescence Versus Stress-Induced Senescence
3. Cellular Senescence in Adult Lungs
3.1. Age-Related (Replicative) Senescence in Adult Lungs
3.2. Cellular Stress-Induced Senescence in Adult Lungs
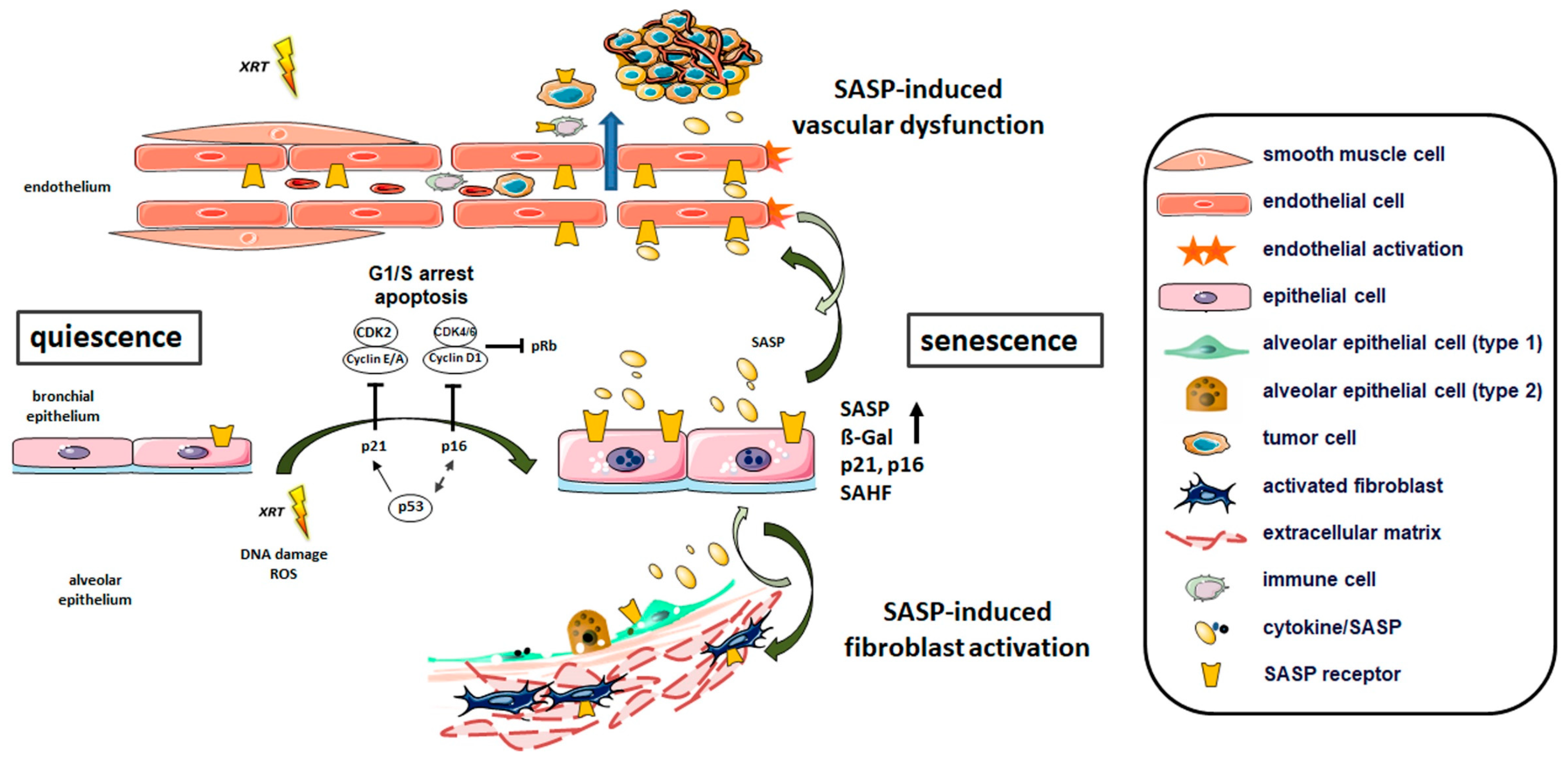
3.2.1. Radiation-Induced Cellular Senescence in Lungs
Senescence of Lung Fibroblasts
Senescence of Lung Endothelial Cells
Senescence of Lung Epithelial Cells
3.2.2. Senescence of Lung Epithelial Cells: Cellular Stressors Other than RT
3.3. Perspective: Biomarker Potential of SASP
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AEC | Alveolar epithelial cell type |
| AP | Activator protein |
| ATM | Ataxia-telangiectasia-mutated |
| ATR | Ataxia-telangiectasia-mutated and Rad3 related |
| BCAA | Branch chain amino acid |
| BLM | Bleomycin |
| BrdU | Bromodeoxyuridine |
| CAF | Cancer associated fibroblast |
| CCL | CC-chemokine ligand |
| CDK | Cyclin-dependent kinase |
| COPD | Chronic obstructive pulmonary disease |
| CST | Cystatin-S |
| CXCL | C-X-C motif ligand |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DDR | DNA damage response |
| DSB | Double-strand breaks |
| ECM | Extracellular matrix |
| ESM | Extracellular senescence metabolome |
| GDF | Growth/ differentiation factor |
| GPX | Glutathione peroxidase |
| HMGB | High mobility group box |
| IGF | Insulin-like growth factor |
| IGFBP | Insulin-like growth factor binding protein |
| IL | Interleukin |
| ILD | Interstitial lung disease |
| IMRT | Intensity-modulated radiotherapy |
| IPF | Idiopathic pulmonary fibrosis |
| IR | Ionizing radiation |
| JAK | Janus kinase |
| LAMB | Lamin subunit beta |
| LPS | Lipopolysaccharide |
| LT | Leukotrien |
| MMP | Matrix metalloprotease |
| MnTBAP | Mn(III) tetrakis (4-benzoic acid) porphyrin |
| mTOR | Mammalian target of rapamycin |
| NFκB | Nuclear factor kappa B |
| NK | Natural killer cell |
| NRF | Nuclear factor (erythroid-derived 2)-like |
| PAI | Plasminogen activator protein |
| PG | Prostagladin |
| PI3K | Phosphoinositide-3-kinase |
| PTEN | Phosphatase tensin homolog |
| P53 | Tumor protein p53, celullar tumor antigen p53 |
| Rb | Retinoblastoma |
| ROS | Reactive oxygen species |
| SA-β-gal | Senescence-associated β-galactosidase |
| SAHF | Senescence-associated heterochromatin foci |
| SASP | Senescence-associated secretory phenotype |
| SBRT | Stereotactic body radiation therapy |
| SERPIN | Serine protease inhibitor |
| SERPINE | Plasminogen activator inhibitor |
| SIPS | Stress-induced premature senescence |
| SOD | Superoxide dismutase |
| TGFβ | Transforming growth factor beta |
| Rb | Retinoblastoma protein |
| RT | Radiotherapy |
| STAT | Signal transducer and activator of transcription |
| STC | Stanniocalcin |
| TERT | Telomerase reverse transcriptase |
| TIMP | Tissue inhibitors of metallopeptidase |
| TR | RNA template |
| XRT | Irradiation |
| ZEB | Zinc finger E-boxing binding homeobox |
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Lundblad, V. The end replication problem: More than one solution. Nat. Med. 1997, 3, 1198–1199. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147 e16. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Ma, X.; Cui, C.; Deenik, P.R.; Henderson, P.K.P.; Sigler, A.L.; Cui, L. Real-time imaging of senescence in tumors with DNA damage. Sci. Rep. 2019, 9, 2102. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Torres, B.; Galiana, I.; Rovira, M.; Garrido, E.; Chaib, S.; Bernardos, A.; Munoz-Espin, D.; Serrano, M.; Martinez-Manez, R.; Sancenon, F. An OFF-ON Two-Photon Fluorescent Probe for Tracking Cell Senescence in Vivo. J. Am. Chem. Soc. 2017, 139, 8808–8811. [Google Scholar] [CrossRef] [PubMed]
- De Keizer, P.L. The Fountain of Youth by Targeting Senescent Cells? Trends Mol. Med. 2017, 23, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Birch, H.L. Extracellular Matrix and Ageing. Subcell Biochem. 2018, 90, 169–190. [Google Scholar]
- Hibbert, S. Extracellular matrix is an influential force in ageing. Br. J. Dermatol. 2017, 177, 1160–1161. [Google Scholar] [CrossRef]
- Prata, L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Sun, Y.; Coppe, J.P.; Lam, E.W. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Wiesemann, A.; Ketteler, J.; Slama, A.; Wirsdorfer, F.; Hager, T.; Rock, K.; Engel, D.R.; Fischer, J.W.; Aigner, C.; Jendrossek, V.; et al. Inhibition of Radiation-Induced Ccl2 Signaling Protects Lungs from Vascular Dysfunction and Endothelial Cell Loss. Antioxid. Redox Signal. 2019, 30, 213–231. [Google Scholar] [CrossRef]
- Klein, D.; Steens, J.; Wiesemann, A.; Schulz, F.; Kaschani, F.; Rock, K.; Yamaguchi, M.; Wirsdorfer, F.; Kaiser, M.; Fischer, J.W.; et al. Mesenchymal Stem Cell Therapy Protects Lungs from Radiation-Induced Endothelial Cell Loss by Restoring Superoxide Dismutase 1 Expression. Antioxid. Redox Signal. 2017, 26, 563–582. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [PubMed]
- Hamsanathan, S.; Alder, J.K.; Sellares, J.; Rojas, M.; Gurkar, A.U.; Mora, A.L. Cellular Senescence: The Trojan Horse in Chronic Lung Diseases. Am. J. Respir Cell Mol. Biol. 2019, 61, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir Crit Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Toussaint, O.; Royer, V.; Salmon, M.; Remacle, J. Stress-induced premature senescence and tissue ageing. Biochem. Pharmacol. 2002, 64, 1007–1009. [Google Scholar] [CrossRef]
- Lozano-Torres, B.; Estepa-Fernández, A.; Rovira, M.; Orzáez, M.; Serrano, M.; Martínez-Máñez, R.; Sancenón, F. The chemistry of senescence. Nat. Rev. Chem. 2019, 3, 426–441. [Google Scholar] [CrossRef]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef]
- Dumont, P.; Burton, M.; Chen, Q.M.; Gonos, E.S.; Frippiat, C.; Mazarati, J.B.; Eliaers, F.; Remacle, J.; Toussaint, O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol. Med. 2000, 28, 361–373. [Google Scholar] [CrossRef]
- Dierick, J.F.; Eliaers, F.; Remacle, J.; Raes, M.; Fey, S.J.; Larsen, P.M.; Toussaint, O. Stress-induced premature senescence and replicative senescence are different phenotypes, proteomic evidence. Biochem. Pharmacol. 2002, 64, 1011–1017. [Google Scholar] [CrossRef]
- Gonos, E.S.; Derventzi, A.; Kveiborg, M.; Agiostratidou, G.; Kassem, M.; Clark, B.F.; Jat, P.S.; Rattan, S.I. Cloning and identification of genes that associate with mammalian replicative senescence. Exp. Cell Res. 1998, 240, 66–74. [Google Scholar] [CrossRef]
- Pascal, T.; Debacq-Chainiaux, F.; Chretien, A.; Bastin, C.; Dabee, A.F.; Bertholet, V.; Remacle, J.; Toussaint, O. Comparison of replicative senescence and stress-induced premature senescence combining differential display and low-density DNA arrays. FEBS Lett. 2005, 579, 3651–3659. [Google Scholar] [CrossRef] [PubMed]
- Kural, K.C.; Tandon, N.; Skoblov, M.; Kel-Margoulis, O.V.; Baranova, A.V. Pathways of aging: Comparative analysis of gene signatures in replicative senescence and stress induced premature senescence. BMC Genom. 2016, 17 (Suppl. 14). [Google Scholar] [CrossRef] [PubMed]
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D., Jr.; Prakash, Y.S. Cellular senescence in the lung across the age spectrum. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L826–L842. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, S.L.; Pruzan, R.; Ma, L.; Ouellette, M.; Tesmer, V.M.; Holt, S.E.; Bodnar, A.G.; Lichtsteiner, S.; Kim, N.W.; Trager, J.B.; et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 1997, 17, 498–502. [Google Scholar] [CrossRef]
- Sun, L.; Chiang, J.Y.; Choi, J.Y.; Xiong, Z.M.; Mao, X.; Collins, F.S.; Hodes, R.J.; Cao, K. Transient induction of telomerase expression mediates senescence and reduces tumorigenesis in primary fibroblasts. Proc. Natl. Acad. Sci. USA 2019, 116, 18983–18993. [Google Scholar] [CrossRef]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Liu, T.; Ullenbruch, M.; Young Choi, Y.; Yu, H.; Ding, L.; Xaubet, A.; Pereda, J.; Feghali-Bostwick, C.A.; Bitterman, P.B.; Henke, C.A.; et al. Telomerase and telomere length in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 260–268. [Google Scholar] [CrossRef]
- Tsang, A.R.; Wyatt, H.D.; Ting, N.S.; Beattie, T.L. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell 2012, 11, 482–490. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, A.; Gomez-Lopez, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife 2018, 7, e31299. [Google Scholar] [CrossRef]
- Liu, T.; Gonzalez De Los Santos, F.; Zhao, Y.; Wu, Z.; Rinke, A.E.; Kim, K.K.; Phan, S.H. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J. Biol. Chem. 2019, 294, 8861–8871. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers. 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.Q.; To, N.H.; Zadigue, P.; Kerbrat, S.; De La Taille, A.; Le Gouvello, S.; Belkacemi, Y. Ionizing radiation-induced cellular senescence promotes tissue fibrosis after radiotherapy. A review. Crit. Rev. Oncol. Hematol. 2018, 129, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Guckenberger, M.; Baier, K.; Polat, B.; Richter, A.; Krieger, T.; Wilbert, J.; Mueller, G.; Flentje, M. Dose-response relationship for radiation-induced pneumonitis after pulmonary stereotactic body radiotherapy. Radiother. Oncol. 2010, 97, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Borst, G.R.; Ishikawa, M.; Nijkamp, J.; Hauptmann, M.; Shirato, H.; Onimaru, R.; van den Heuvel, M.M.; Belderbos, J.; Lebesque, J.V.; Sonke, J.J. Radiation pneumonitis in patients treated for malignant pulmonary lesions with hypofractionated radiation therapy. Radiother. Oncol. 2009, 91, 307–313. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Thummuri, D.; Zheng, G.; Okunieff, P.; Citrin, D.E.; Vujaskovic, Z.; Zhou, D. Cellular senescence and radiation-induced pulmonary fibrosis. Transl Res. 2019, 209, 14–21. [Google Scholar] [CrossRef]
- Kelsey, C.R.; Horwitz, M.E.; Chino, J.P.; Craciunescu, O.; Steffey, B.; Folz, R.J.; Chao, N.J.; Rizzieri, D.A.; Marks, L.B. Severe pulmonary toxicity after myeloablative conditioning using total body irradiation: An assessment of risk factors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 812–818. [Google Scholar] [CrossRef]
- Down, J.D.; Yanch, J.C. Identifying the high radiosensitivity of the lungs of C57L mice in a model of total-body irradiation and bone marrow transplantation. Radiat. Res. 2010, 174, 258–263. [Google Scholar] [CrossRef]
- Koukourakis, M.I. Radiation damage and radioprotectants: new concepts in the era of molecular medicine. Br. J. Radiol. 2012, 85, 313–330. [Google Scholar] [CrossRef]
- Bentzen, S.M. Preventing or reducing late side effects of radiation therapy: Radiobiology meets molecular pathology. Nat. Rev. Cancer 2006, 6, 702–713. [Google Scholar] [CrossRef]
- Thompson, M.; Rosenzweig, K.E. The evolving toxicity profile of SBRT for lung cancer. Transl. Lung Cancer Res. 2019, 8, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.; Dragojevic, I.; Hoisak, J.; Hoopes, D.; Manger, R. Lung Stereotactic Body Radiation Therapy (SBRT) dose gradient and PTV volume: A retrospective multi-center analysis. Radiat. Oncol. 2019, 14, 162. [Google Scholar] [CrossRef] [PubMed]
- Montay-Gruel, P.; Petersson, K.; Jaccard, M.; Boivin, G.; Germond, J.F.; Petit, B.; Doenlen, R.; Favaudon, V.; Bochud, F.; Bailat, C.; et al. Irradiation in a flash: Unique sparing of memory in mice after whole brain irradiation with dose rates above 100Gy/s. Radiother Oncol. 2017, 124, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Fouillade, C.; Curras-Alonso, S.; Giuranno, L.; Quelennec, E.; Heinrich, S.; Bonnet-Boissinot, S.; Beddok, A.; Leboucher, S.; Karakurt, H.U.; Bohec, M.; et al. FLASH Irradiation Spares Lung Progenitor Cells and Limits the Incidence of Radio-induced Senescence. Clin. Cancer Res. 2019, 26, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Soysouvanh, F.; Benadjaoud, M.A.; Dos Santos, M.; Mondini, M.; Lavigne, J.; Bertho, A.; Buard, V.; Tarlet, G.; Adnot, S.; Deutsch, E.; et al. Stereotactic Lung Irradiation in Mice Promotes Long-Term Senescence and Lung Injury. Int. J. Radiat Oncol. Biol. Phys. 2020, 106, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir Crit Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef]
- Xie, T.; Wang, Y.; Deng, N.; Huang, G.; Taghavifar, F.; Geng, Y.; Liu, N.; Kulur, V.; Yao, C.; Chen, P.; et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep. 2018, 22, 3625–3640. [Google Scholar] [CrossRef]
- White, E.S. Lung extracellular matrix and fibroblast function. Ann. Am. Thorac Soc. 2015, 12 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef]
- Barron, L.; Gharib, S.A.; Duffield, J.S. Lung Pericytes and Resident Fibroblasts: Busy Multitaskers. Am. J. Pathol. 2016, 186, 2519–2531. [Google Scholar] [CrossRef]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Di Carlo, S.E.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, Z.; Ning, W. Advances in Molecular Mechanisms and Treatment of Radiation-Induced Pulmonary Fibrosis. Transl. Oncol. 2019, 12, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Hou, J.; Wang, H.; Zheng, Y.; Li, H.; Cai, H.; Han, X.; Dai, J. Epithelial cell senescence induces pulmonary fibrosis through Nanog-mediated fibroblast activation. Aging (Albany NY) 2019, 12, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Kletsas, D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int. J. Oncol. 2011, 39, 989–999. [Google Scholar] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Jing, H.; Lee, S. NF-kappaB in cellular senescence and cancer treatment. Mol. Cells 2014, 37, 189–195. [Google Scholar] [CrossRef]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-associated fibroblasts in radiotherapy: Challenges and new opportunities. Cell Commun. Signal. CCS 2019, 17, 47. [Google Scholar] [CrossRef]
- Aliper, A.M.; Bozdaganyan, M.E.; Orekhov, P.S.; Zhavoronkov, A.; Osipov, A.N. Replicative and radiation-induced aging: A comparison of gene expression profiles. Aging (Albany NY) 2019, 11, 2378–2387. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef] [PubMed]
- Baselet, B.; Sonveaux, P.; Baatout, S.; Aerts, A. Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cell Mol. Life Sci. 2019, 76, 699–728. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef]
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.C.; Gaugler, M.H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic Biol. Med. 2017, 108, 750–759. [Google Scholar] [CrossRef]
- Aratani, S.; Tagawa, M.; Nagasaka, S.; Sakai, Y.; Shimizu, A.; Tsuruoka, S. Radiation-induced premature cellular senescence involved in glomerular diseases in rats. Sci. Rep. 2018, 8, 16812. [Google Scholar] [CrossRef]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef]
- Hellweg, C.E. The Nuclear Factor kappaB pathway: A link to the immune system in the radiation response. Cancer Lett. 2015, 368, 275–289. [Google Scholar] [CrossRef]
- Dong, X.; Tong, F.; Qian, C.; Zhang, R.; Dong, J.; Wu, G.; Hu, Y. NEMO modulates radiation-induced endothelial senescence of human umbilical veins through NF-kappaB signal pathway. Radiat Res. 2015, 183, 82–93. [Google Scholar] [CrossRef]
- Sabatino, L.; Picano, E.; Andreassi, M.G. Telomere shortening and ionizing radiation: A possible role in vascular dysfunction? Int. J. Radiat Biol. 2012, 88, 830–839. [Google Scholar] [CrossRef]
- Igarashi, K.; Miura, M. Inhibition of a radiation-induced senescence-like phenotype: A possible mechanism for potentially lethal damage repair in vascular endothelial cells. Radiat Res. 2008, 170, 534–539. [Google Scholar] [CrossRef][Green Version]
- Yanagi, S.; Tsubouchi, H.; Miura, A.; Matsuo, A.; Matsumoto, N.; Nakazato, M. The Impacts of Cellular Senescence in Elderly Pneumonia and in Age-Related Lung Diseases That Increase the Risk of Respiratory Infections. Int. J. Mol. Sci. 2017, 18, 503. [Google Scholar] [CrossRef]
- Yanagihara, T.; Sato, S.; Upagupta, C.; Kolb, M. What have we learned from basic science studies on idiopathic pulmonary fibrosis? Eur. Respir. Rev. 2019, 28, 190029. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir J. 2017, 50, 1602367. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat Oncol. Biol. Phys. 2017, 99, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Schmetter, A.; Imsak, R.; Wirsdorfer, F.; Unger, K.; Jastrow, H.; Stuschke, M.; Jendrossek, V. Therapy with Multipotent Mesenchymal Stromal Cells Protects Lungs from Radiation-Induced Injury and Reduces the Risk of Lung Metastasis. Antioxid. Redox Signal. 2016, 24, 53–69. [Google Scholar] [CrossRef]
- Wunderlich, R.; Ruehle, P.F.; Deloch, L.; Unger, K.; Hess, J.; Zitzelsberger, H.; Lauber, K.; Frey, B.; Gaipl, U.S. Interconnection between DNA damage, senescence, inflammation, and cancer. Front. BioSci. (Landmark Ed.) 2017, 22, 348–369. [Google Scholar]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Kuznar-Kaminska, B.; Mikula-Pietrasik, J.; Witucka, A.; Romaniuk, A.; Konieczna, N.; Rubis, B.; Ksiazek, K.; Tykarski, A.; Batura-Gabryel, H. Serum from patients with chronic obstructive pulmonary disease induces senescence-related phenotype in bronchial epithelial cells. Sci. Rep. 2018, 8, 12940. [Google Scholar] [CrossRef]
- Bodas, M.; Van Westphal, C.; Carpenter-Thompson, R.; Mohanty, D.K.; Vij, N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xuan, W.; Wang, H.; Sun, F.; Zhang, C.; Gong, Q.; Ge, S. miR-200b regulates cellular senescence and inflammatory responses by targeting ZEB2 in pulmonary emphysema. Artif. Cells Nanomed Biotechnol. 2020, 48, 656–663. [Google Scholar] [CrossRef]
- Sagiv, A.; Bar-Shai, A.; Levi, N.; Hatzav, M.; Zada, L.; Ovadya, Y.; Roitman, L.; Manella, G.; Regev, O.; Majewska, J.; et al. p53 in Bronchial Club Cells Facilitates Chronic Lung Inflammation by Promoting Senescence. Cell Rep. 2018, 22, 3468–3479. [Google Scholar] [CrossRef] [PubMed]
- James, E.N.L.; Bennett, M.H.; Parkinson, E.K. The induction of the fibroblast extracellular senescence metabolome is a dynamic process. Sci. Rep. 2018, 8, 12148. [Google Scholar] [CrossRef] [PubMed]
- Von Kobbe, C. Targeting senescent cells: Approaches, opportunities, challenges. Aging 2019, 11, 12844–12861. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Mausset-Bonnefont, A.L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.P.; Effros, R.B. T cell replicative senescence in human aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K., Jr.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Cahu, J.; Sola, B. A sensitive method to quantify senescent cancer cells. J. Vis. Exp. 2013, 78, e50494. [Google Scholar] [CrossRef]
- Hennel, R.; Brix, N.; Seidl, K.; Ernst, A.; Scheithauer, H.; Belka, C.; Lauber, K. Release of monocyte migration signals by breast cancer cell lines after ablative and fractionated γ-irradiation. Radiat. Oncol. (Lond. Engl.) 2014, 9, 85. [Google Scholar] [CrossRef]
- Evangelou, K.; Gorgoulis, V.G. Sudan Black B, The Specific Histochemical Stain for Lipofuscin: A Novel Method to Detect Senescent Cells. Methods Mol. Biol. 2017, 1534, 111–119. [Google Scholar]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef]
- Ourliac-Garnier, I.; Londono-Vallejo, A. Telomere length analysis by quantitative fluorescent in situ hybridization (Q-FISH). Methods Mol. Biol. 2011, 735, 21–31. [Google Scholar]
- Martens, U.M.; Brass, V.; Engelhardt, M.; Glaser, S.; Waller, C.F.; Lange, W.; Schmoor, C.; Poon, S.S.; Landsdorp, P.M. Measurement of telomere length in haematopoietic cells using in situ hybridization techniques. Biochem. Soc. Trans. 2000, 28, 245–250. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196. [Google Scholar]


{kind=link}
| Target | Marker | Method of Detection | |
|---|---|---|---|
| Lysosomes | SA-β-gal | Histochemical detection of β-galactosidase activity at pH 6 [9,20,22,23,24,25,26,27,28,29] | |
| Fluorogenic probes (e.g., C12FDG) [30,31] | |||
| Near-infrared molecular probe (in vivo and in vitro) [32] | |||
| Two-photon fluorescent probe (in vivo and in vitro) [33] | |||
| Lipofuscin | Lysosomal aggregates stained with Sudan Black B [34] | ||
| Cell cycle inhibitors | p16INK4a, p21Cip/Waf1, p15INK4b, p27 | Western blot [9,18,19,20,23,24,29,35] | |
| RT-PCR [20,35,36,37] | |||
| Immunofluorescence [35,38] | |||
| Immunohistochemistry [29,39] | |||
| Cell proliferation | Ki-67 (absence) | Western Blot [40] | |
| RT-PCR [40] | |||
| Immunofluorescence [38] | |||
| BrdU incorporation (absence) | Immunofluorescence [18] | ||
| Telomere shortening | FISH [41,42] | ||
| SASP factors | Cytokines (e.g., IL-6, TNFα) Chemokines (e.g., IL-8, MIPs, CCLs) Proteases (e.g., MMPs) Candidates: TGFβ, GM-CSF, PAI-1, IGF-1 | Immunofluorescence [19,20] | |
| RT-PCR [9,20,25,26,37,38,43] | |||
| Western Blot [9,19,20] | |||
| Tumor suppressors | pPTEN, p53, hypo-phosphorylated Rb, FOXO4 | Western blot [9,18,24] | |
| RT-PCR [36] | |||
| Immunofluorescence [6] | |||
| Chromatin organization | SAHF | NFκB p65 subunit | Immunofluorescence [9] |
| Western Blot [9] | |||
| RT-PCR [9] | |||
| reorganization of DNA structure by DAPI, antibodies against facultative heterochromatin | Immunofluorescence [44] | ||
| DNA damage marker | γH2AX | Western blot [45] | |
| Immunofluorescence [27,28] | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansel, C.; Jendrossek, V.; Klein, D. Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3279. https://doi.org/10.3390/ijms21093279
Hansel C, Jendrossek V, Klein D. Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(9):3279. https://doi.org/10.3390/ijms21093279
Chicago/Turabian StyleHansel, Christine, Verena Jendrossek, and Diana Klein. 2020. "Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells" International Journal of Molecular Sciences 21, no. 9: 3279. https://doi.org/10.3390/ijms21093279
APA StyleHansel, C., Jendrossek, V., & Klein, D. (2020). Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells. International Journal of Molecular Sciences, 21(9), 3279. https://doi.org/10.3390/ijms21093279