Highly Expressed FOXF1 Inhibit Non-Small-Cell Lung Cancer Growth via Inducing Tumor Suppressor and G1-Phase Cell-Cycle Arrest

 , and
, and 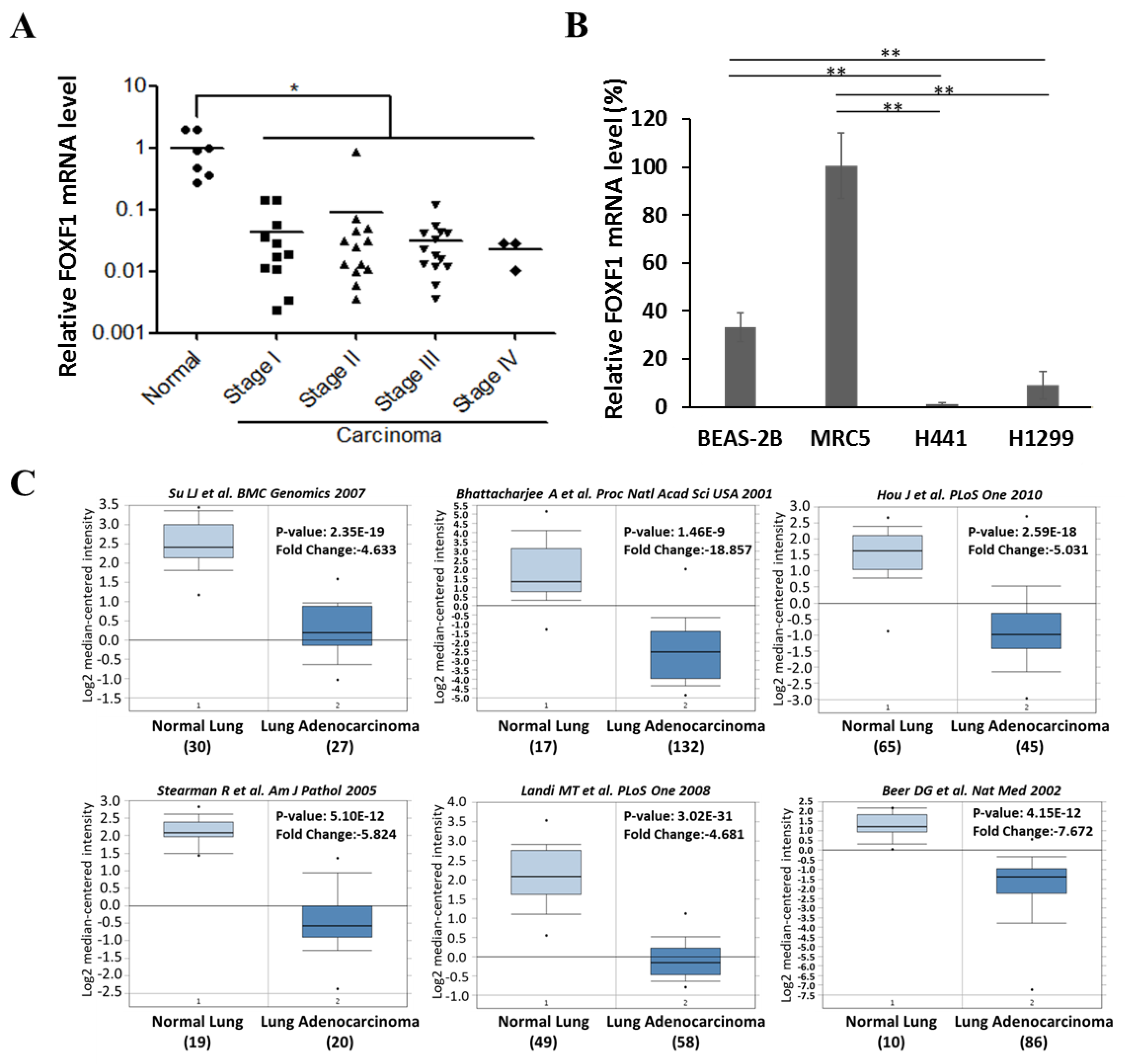{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. FOXF1 is Downregulated in Lung Cancer Tissue and Cell Lines
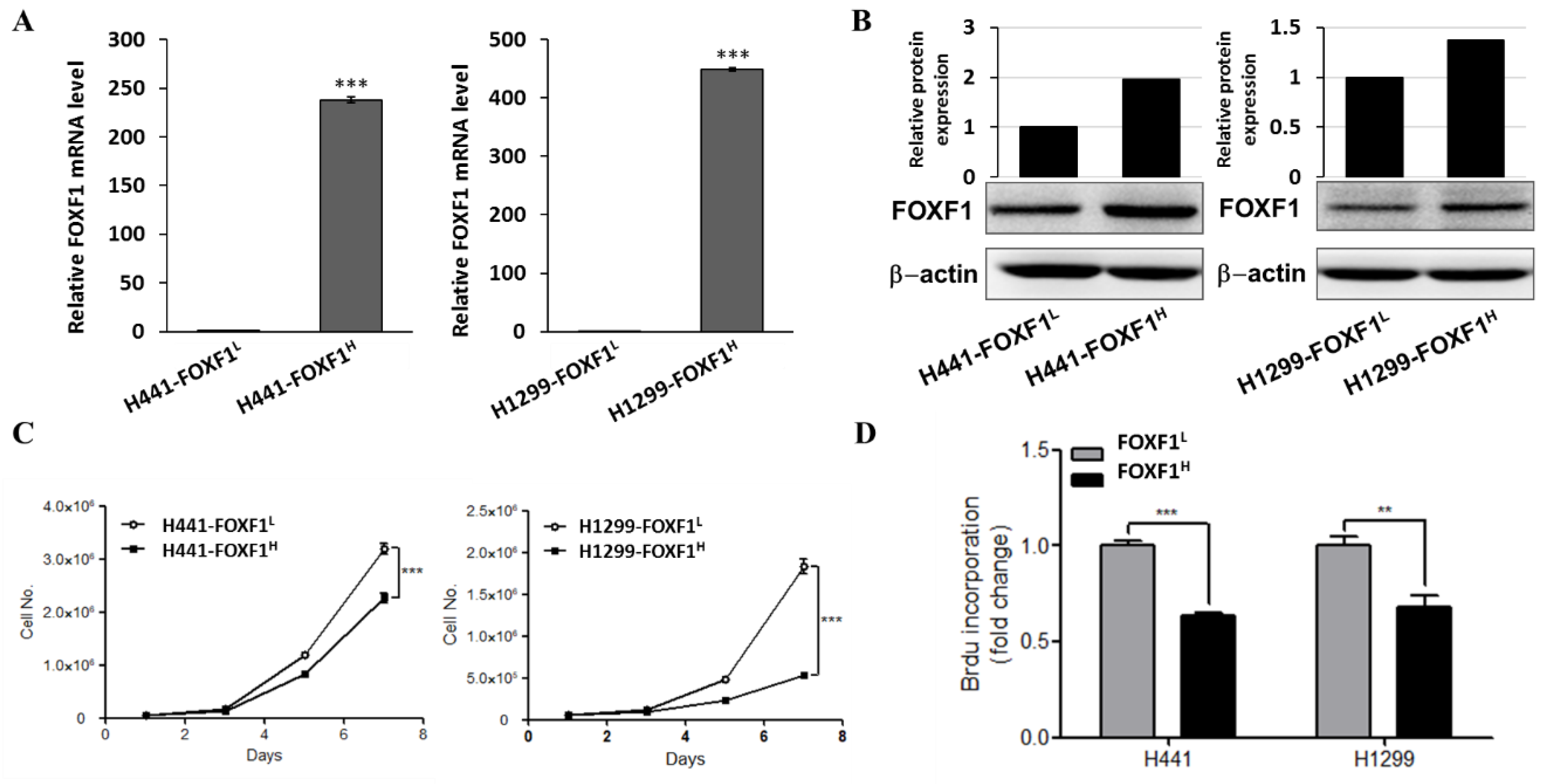
2.2. Highly Expressed FOXF1 Lung Cancer Cell Showed Inhibited Cell Proliferation Ability
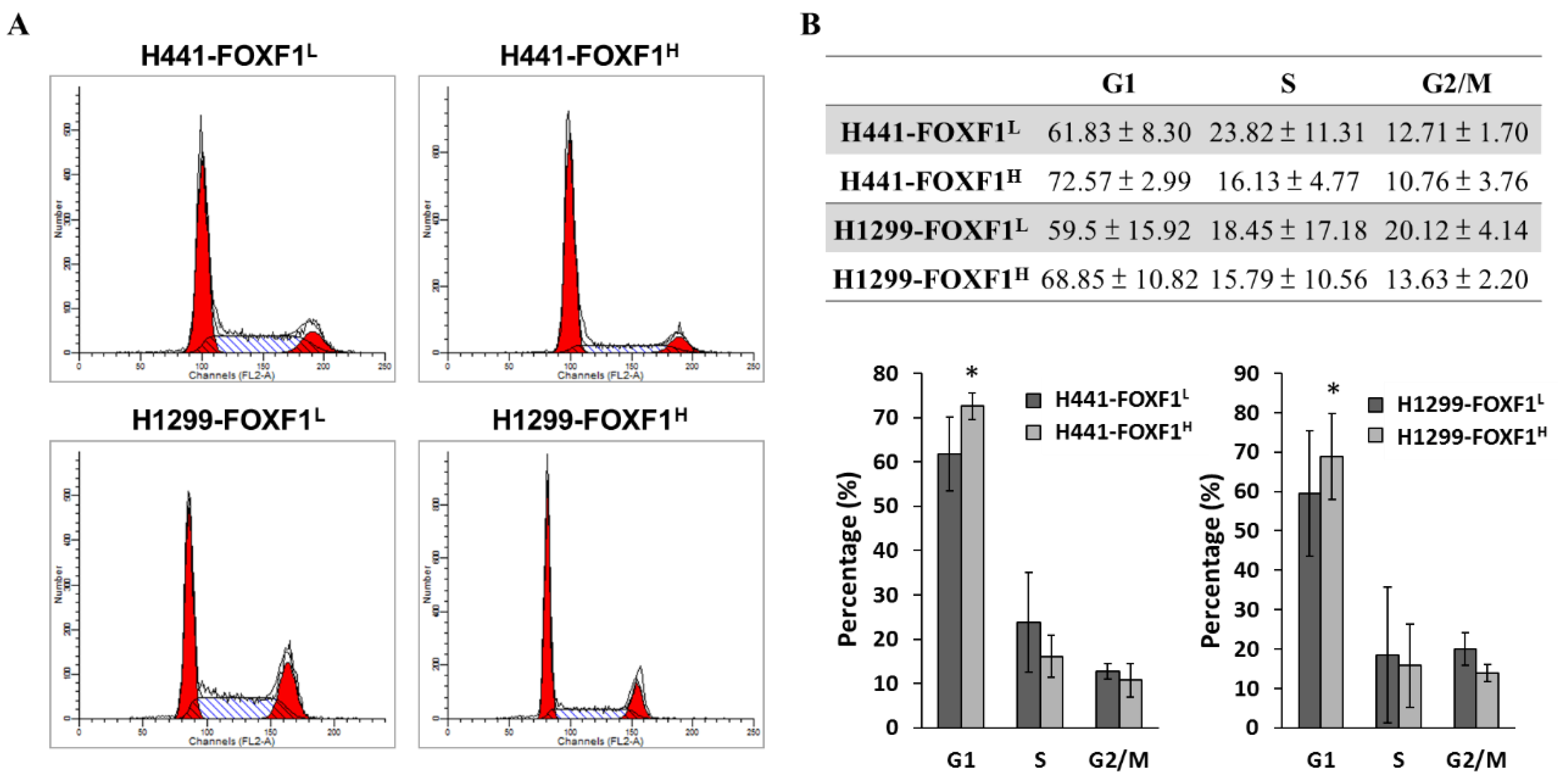
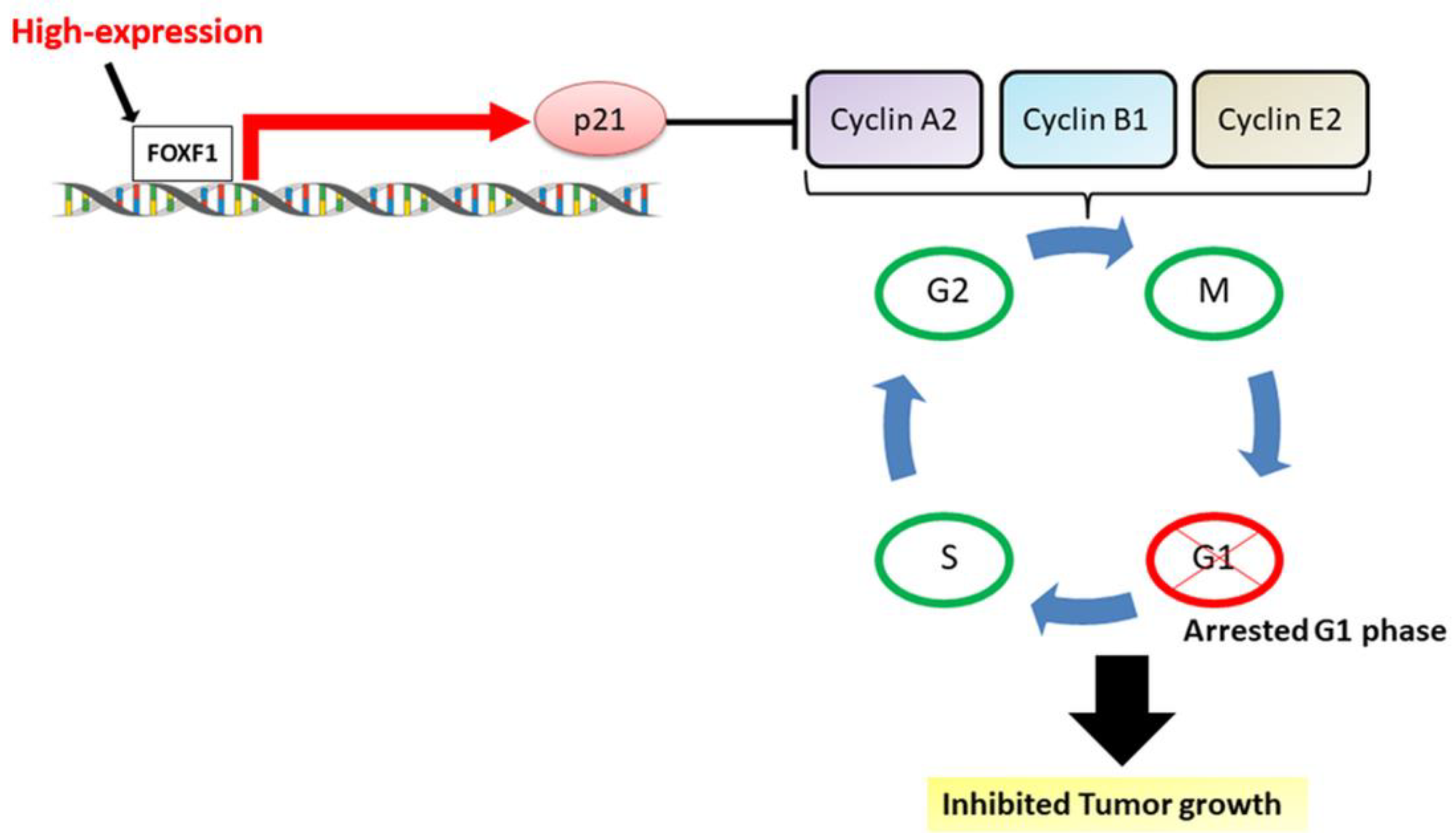
2.3. Highly Expressed FOXF1 Promotes G1 Cell-Cycle Arrest
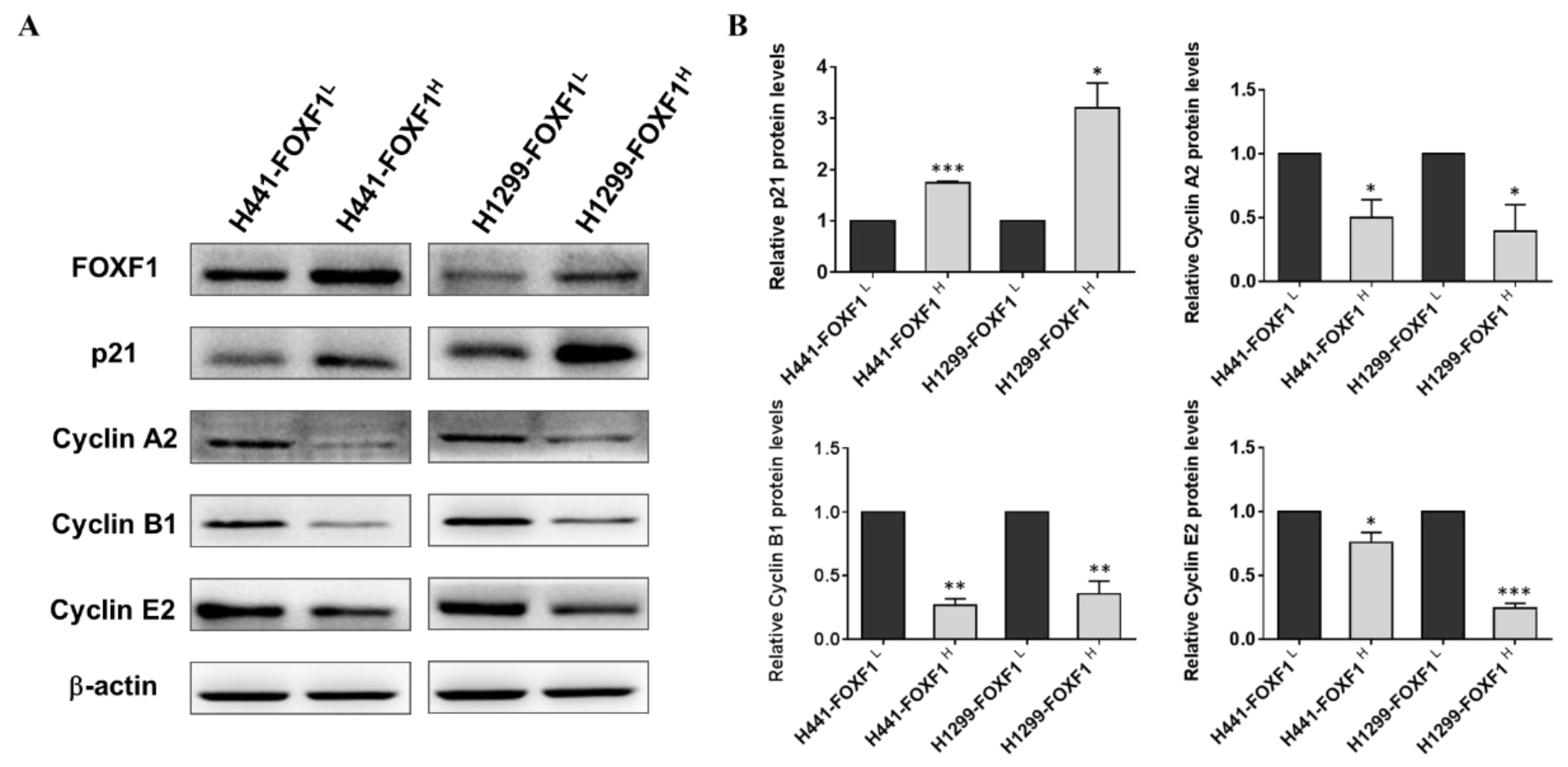
2.4. High Expression of FOXF1 Promotes Tumor Suppression and Inhibits Cellular Cyclins
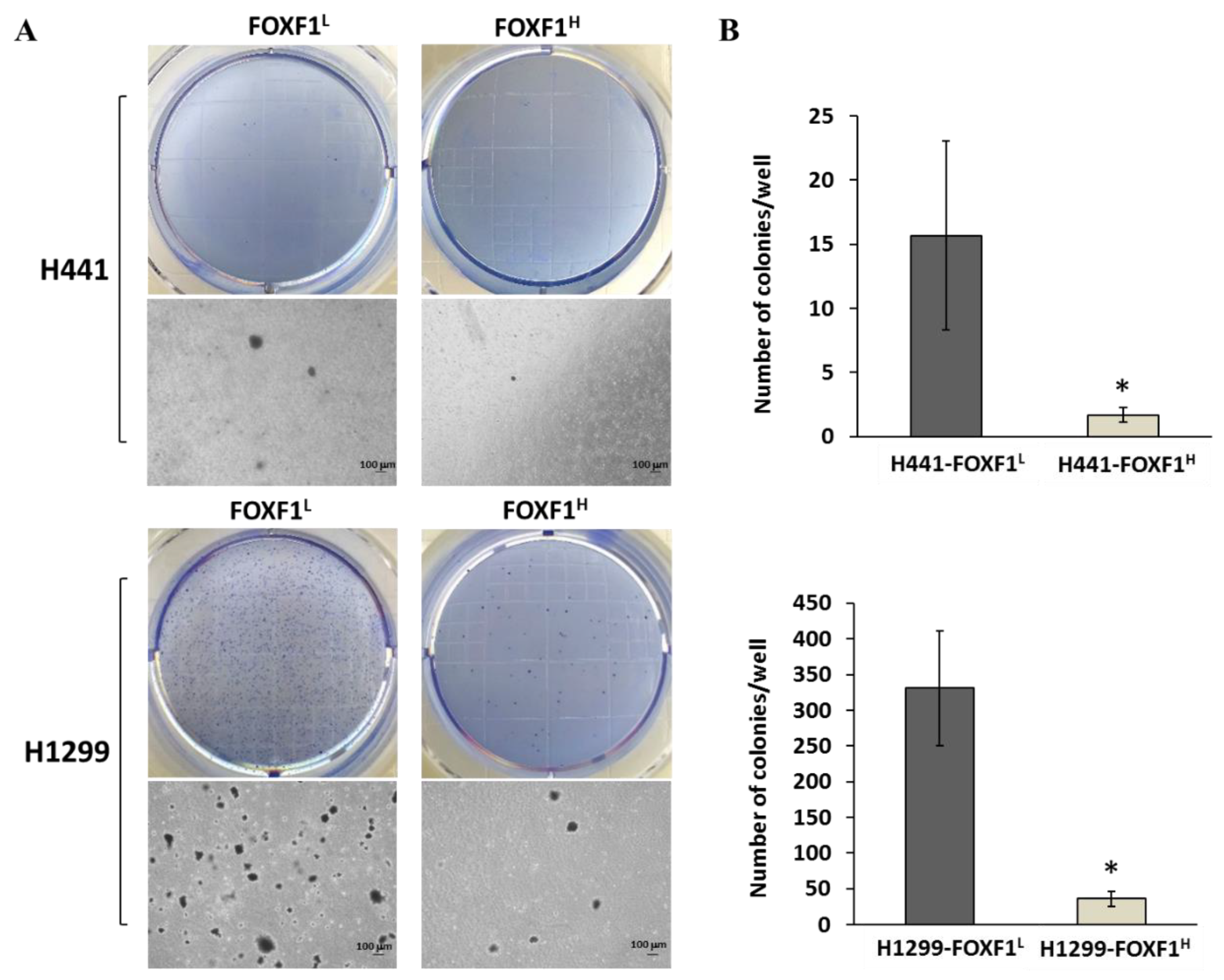
2.5. High Expression of FOXF1 Inhibits Anchorage-Independent Growth Ability and Transformation Ability
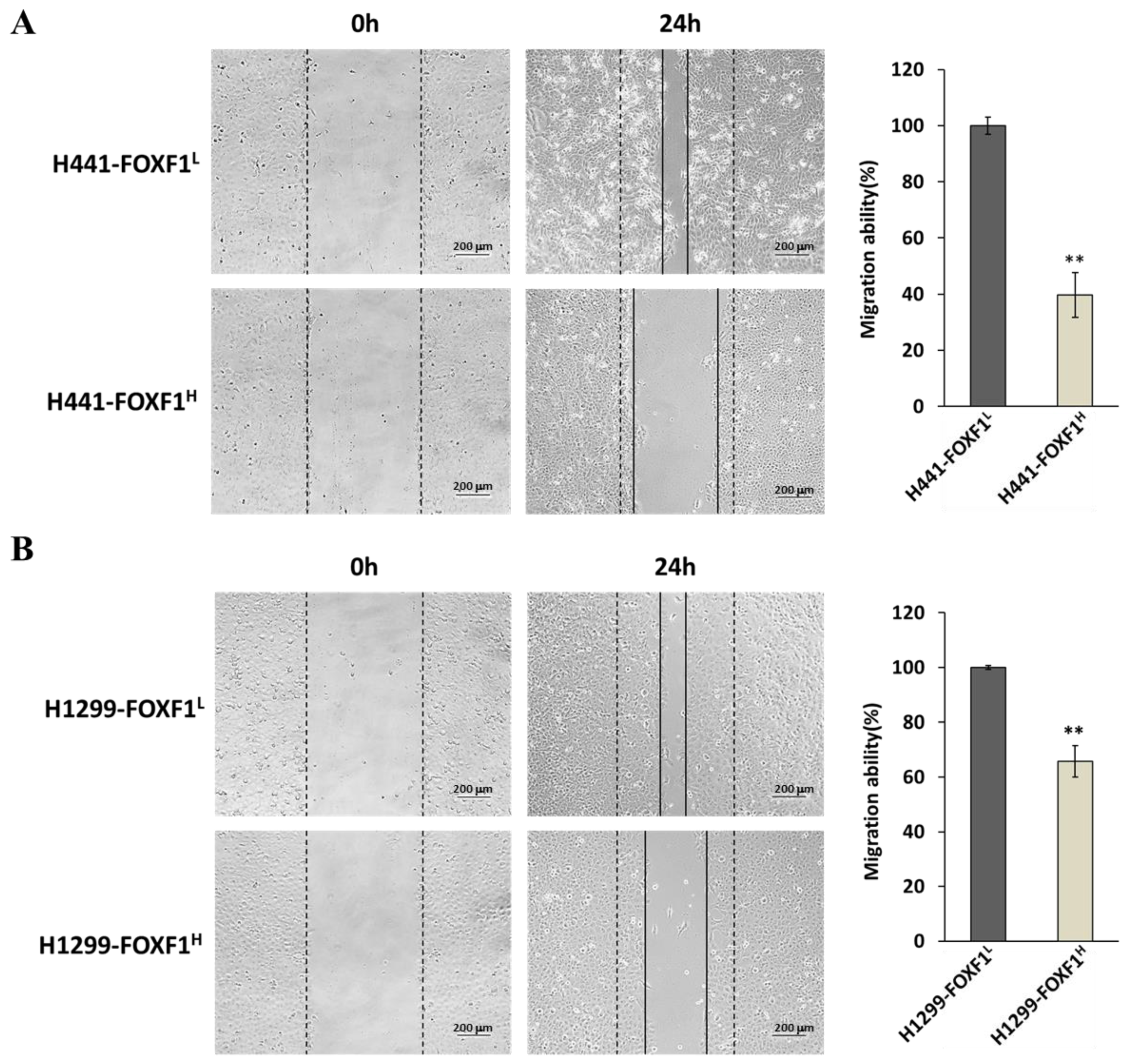
2.6. High Expression of FOXF1 Inhibits Lung Cancer Cell Migration Ability
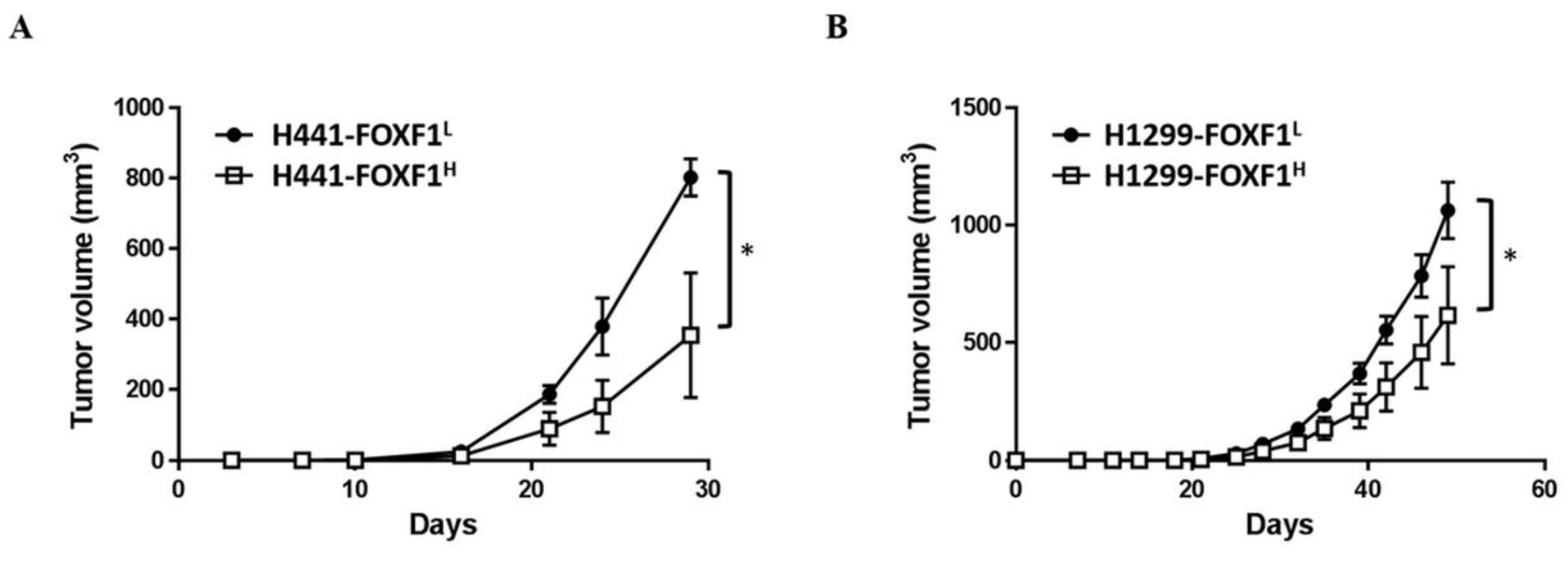
2.7. High Expression of FOXF1 Inhibits Lung Cancer Cell Tumorigenicity In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animal Studies
4.3. In Silico Analysis of FOXF1 Gene Expression
4.4. Higher Expression of FOXF1
4.5. Real-Time Polymerase Chain Reaction (PCR)
4.6. Western Blot Analysis
4.7. Cell Count
4.8. Cell Proliferation Assay
4.9. Cell-Cycle Analysis
4.10. Anchorage-Independent Growth
4.11. Wound Healing Assay
4.12. Tumorigenicity In Vivo
4.13. Statistical Analysis and Replicates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FOXF1 | Forkhead box F1 |
| NSCLC | Non-small-cell lung cancer |
References
- Henley, S.J.; Gallaway, S.; Singh, S.D.; O’Neil, M.E.; Buchanan Lunsford, N.; Momin, B.; Richards, T.B. Lung cancer among women in the united states. J. Womens Health 2018, 27, 1307–1316. [Google Scholar] [CrossRef]
- Kuo, C.N.; Liao, Y.M.; Kuo, L.N.; Tsai, H.J.; Chang, W.C.; Yen, Y. Cancers in taiwan: Practical insight from epidemiology, treatments, biomarkers, and cost. J. Med. Assoc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, F.; Ebrahimi, M.; Goliaei, B.; Shamabadi, N.J.P.o. Classification of lung cancer tumors based on structural and physicochemical properties of proteins by bioinformatics models. PLoS ONE 2012, 7, e40017. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Tanaka, F. Recurrence after surgery in patients with nsclc. Transl. Lung Cancer Res. 2014, 3, 242. [Google Scholar]
- Al-Kattan, K.; Sepsas, E.; Fountain, S.W.; Townsend, E.R. Disease recurrence after resection for stage i lung cancer. Eur. J. Cardio-Thorac. Surg. 1997, 12, 380–384. [Google Scholar] [CrossRef]
- Hoffman, P.C.; Mauer, A.M.; Vokes, E.E. Lung cancer. Lancet 2000, 355, 479–485. [Google Scholar] [CrossRef]
- Albain, K.S.; Swann, R.S.; Rusch, V.W.; Turrisi, A.T., III; Shepherd, F.A.; Smith, C.; Chen, Y.; Livingston, R.B.; Feins, R.H.; Gandara, D.R.J.T.L. Radiotherapy plus chemotherapy with or without surgical resection for stage iii non-small-cell lung cancer: A phase iii randomised controlled trial. Lancet 2009, 374, 379–386. [Google Scholar] [CrossRef]
- Thomas, M.; Rübe, C.; Hoffknecht, P.; Macha, H.N.; Freitag, L.; Linder, A.; Willich, N.; Hamm, M.; Sybrecht, G.W.; Ukena, D.J.T.l.o. Effect of preoperative chemoradiation in addition to preoperative chemotherapy: A randomised trial in stage iii non-small-cell lung cancer. Lancet Oncol. 2008, 9, 636–648. [Google Scholar] [CrossRef]
- Pless, M.; Stupp, R.; Ris, H.-B.; Stahel, R.A.; Weder, W.; Thierstein, S.; Xyrafas, A.; Frueh, M.; Cathomas, R.; Zippelius, A. Neoadjuvant Chemotherapy with or Without Preoperative Irradiation in Stage iiia/n2 Non-Small CELL lung Cancer (Nsclc): A Randomized Phase iii Trial by the Swiss Group for Clinical Cancer Research (Sakk Trial 16/00). American Society of Clinical Oncology. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT00030771 (accessed on 9 April 2014).
- Hellqvist, M.; Mahlapuu, M.; Samuelsson, L.; Enerbäck, S.; Carlsson, P.J.J.o.B.C. Differential activation of lung-specific genes by two forkhead proteins, freac-1 and freac-2. J. Biol. Chem. 1996, 271, 4482–4490. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Ormestad, M.; Enerback, S.; Carlsson, P.J.D. The forkhead transcription factor foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development 2001, 128, 155–166. [Google Scholar]
- Stankiewicz, P.; Sen, P.; Bhatt, S.S.; Storer, M.; Xia, Z.; Bejjani, B.A.; Ou, Z.; Wiszniewska, J.; Driscoll, D.J.; Bolivar, J.J.T.A.J.o.H.G. Genomic and genic deletions of the fox gene cluster on 16q24. 1 and inactivating mutations of foxf1 cause alveolar capillary dysplasia and other malformations. Am. J. Hum. Genet. 2009, 84, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Luk, H.M.; Tang, T.; Choy, K.W.R.; Tong, M.F.T.; Wong, O.K.; Lo, F.M.I. Maternal somatic mosaicism of foxf1 mutation causes recurrent alveolar capillary dysplasia with misalignment of pulmonary veins in siblings. Am. J. Med. Genet. Part. A 2016, 170, 1942–1944. [Google Scholar] [CrossRef] [PubMed]
- Alsina Casanova, M.; Monteagudo-Sánchez, A.; Rodiguez Guerineau, L.; Court, F.; Gazquez Serrano, I.; Martorell, L.; Rovira Zurriaga, C.; Moore, G.E.; Ishida, M.; Castañon, M. Maternal mutations of foxf1 cause alveolar capillary dysplasia despite not being imprinted. Hum. Mutat. 2017, 38, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.; Leung, A.; Kwok, C.; Cheung, W.; Ko, J.; Yang, L.; Law, S.; Wang, L.; Li, J.; Stanbridge, E.J.; et al. Identification of a tumor suppressive critical region mapping to 3p14. 2 in esophageal squamous cell carcinoma and studies of a candidate tumor suppressor gene, adamts9. Oncogene 2007, 26, 148–157. [Google Scholar] [CrossRef]
- Tokino, T.; Tamura, M.; Idogawa, M.; Sasaki, Y. Forkhead transcription factor foxf1 is a novel p53 target gene and regulates cancer cell migration and invasion. In Proceedings of the AACR Annual Meeting, San Diego, CA, USA, 5–9 April 2014; p. 598. [Google Scholar]
- Watson, J.V.; Doggett, N.A.; Albertson, D.G.; Andaya, A.; Chinnaiyan, A.; Van Dekken, H.; Ginzinger, D.; Haqq, C.; James, K.; Kamkar, S. Integration of high-resolution array comparative genomic hybridization analysis of chromosome 16q with expression array data refines common regions of loss at 16q23–qter and identifies underlying candidate tumor suppressor genes in prostate cancer. Oncogene 2004, 23, 3487. [Google Scholar] [CrossRef][Green Version]
- Lo, P.-K.; Lee, J.S.; Liang, X.; Han, L.; Mori, T.; Fackler, M.J.; Sadik, H.; Argani, P.; Pandita, T.K.; Sukumar, S. Epigenetic inactivation of the potential tumor suppressor gene foxf1 in breast cancer. Cancer Res. 2010, 70, 6047–6058. [Google Scholar] [CrossRef]
- Wei, H.-J.; Nickoloff, J.A.; Chen, W.-H.; Liu, H.-Y.; Lo, W.-C.; Chang, Y.-T.; Yang, P.-C.; Wu, C.-W.; Williams, D.F.; Gelovani, J.G.; et al. Foxf1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 2014, 5, 9514. [Google Scholar] [CrossRef]
- Su, L.-J.; Chang, C.-W.; Wu, Y.-C.; Chen, K.-C.; Lin, C.-J.; Liang, S.-C.; Lin, C.-H.; Whang-Peng, J.; Hsu, S.-L.; Chen, C.-H.F. Selection of ddx5 as a novel internal control for q-rt-pcr from microarray data using a block bootstrap re-sampling scheme. BMC Genom. 2007, 8, 140. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M. Classification of human lung carcinomas by mrna expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef]
- Hou, J.; Aerts, J.; Den Hamer, B.; Van Ijcken, W.; Den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Stearman, R.S.; Dwyer-Nield, L.; Zerbe, L.; Blaine, S.A.; Chan, Z.; Bunn Jr, P.A.; Johnson, G.L.; Hirsch, F.R.; Merrick, D.T.; Franklin, W.A. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am. J. Pathol. 2005, 167, 1763–1775. [Google Scholar] [CrossRef]
- Landi, M.T.; Dracheva, T.; Rotunno, M.; Figueroa, J.D.; Liu, H.; Dasgupta, A.; Mann, F.E.; Fukuoka, J.; Hames, M.; Bergen, A.W. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 2008, 3, e1651. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.G.; Kardia, S.L.; Huang, C.-C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816. [Google Scholar] [CrossRef] [PubMed]
- Baldi, A.; De Luca, A.; Esposito, V.; Campioni, M.; Spugnini, E.P.; Citro, G. Tumor suppressors and cell-cycle proteins in lung cancer. Pathol. Res. Int. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Velez, A.M.A.; Howard, M.S. Tumor-suppressor genes, cell cycle regulatory checkpoints, and the skin. N. Am. J. Med. Sci. 2015, 7, 176. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Enerbäck, S.; Carlsson, P. Haploinsufficiency of the forkhead gene foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 2001, 128, 2397–2406. [Google Scholar]
- Madison, B.B.; McKenna, L.B.; Dolson, D.; Epstein, D.J.; Kaestner, K.H. Foxf1 and foxl1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J. Biol. Chem. 2009, 284, 5936–5944. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and mirna. Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.S.; Lück, C.; Von Schweinitz, D.; Kappler, R. Characteristic overexpression of the forkhead box transcription factor foxf1 in patched-associated tumors. Int. J. Mol. Med. 2008, 22, 787–792. [Google Scholar] [PubMed]
- Lae, M.; Ahn, E.; Mercado, G.; Chuai, S.; Edgar, M.; Pawel, B.; Olshen, A.; Barr, F.; Ladanyi, M. Global gene expression profiling of pax-fkhr fusion-positive alveolar and pax-fkhr fusion-negative embryonal rhabdomyosarcomas. J. Pathol. 2007, 212, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.-K.; Lee, J.S.; Chen, H.; Reisman, D.; Berger, F.G.; Sukumar, S. Cytoplasmic mislocalization of overexpressed foxf1 is associated with the malignancy and metastasis of colorectal adenocarcinomas. Exp. Mol. Pathol. 2013, 94, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Fulford, L.; Milewski, D.; Ustiyan, V.; Ravishankar, N.; Cai, Y.; Le, T.; Masineni, S.; Kasper, S.; Aronow, B.; Kalinichenko, V.V.; et al. The transcription factor foxf1 promotes prostate cancer by stimulating the mitogen-activated protein kinase erk5. Sci. Signal. 2016, 9, ra48. [Google Scholar] [CrossRef]
- Gialmanidis, I.P.; Bravou, V.; Petrou, I.; Kourea, H.; Mathioudakis, A.; Lilis, I.; Papadaki, H. Expression of bmi1, foxf1, nanog, and γ-catenin in relation to hedgehog signaling pathway in human non-small-cell lung cancer. Lung 2013, 191, 511–521. [Google Scholar] [CrossRef]
- Herrera-Merchan, A.; Cuadros, M.; Rodriguez, M.I.; Rodriguez, S.; Torres, R.; Estecio, M.; Coira, I.F.; Loidi, C.; Saiz, M.; Carmona-Saez, P.; et al. The value of lncrna fendrr and foxf1 as a prognostic factor for survival of lung adenocarcinoma. Oncotarget 2019, 5, 1172–1185. [Google Scholar] [CrossRef]
- Miao, L.; Huang, Z.; Zengli, Z.; Li, H.; Chen, Q.; Yao, C.; Cai, H.; Xiao, Y.; Xia, H.; Wang, Y. Loss of long noncoding rna foxf1-as1 regulates epithelial-mesenchymal transition, stemness and metastasis of non-small cell lung cancer cells. Oncotarget 2016, 7, 68339. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. P21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef]
- Galderisi, U.; Jori, F.P.; Giordano, A. Cell cycle regulation and neural differentiation. Oncogene 2003, 22, 5208–5219. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Adhami, V.M.; Afaq, F.; Feyes, D.K.; Mukhtar, H. Resveratrol causes waf-1/p21-mediated g1-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma a431 cells. Clin. Cancer Res. 2001, 7, 1466–1473. [Google Scholar] [PubMed]
- Collins, K.; Jacks, T.; Pavletich, N.P. The cell cycle and cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 2776–2778. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.; El-Deiry, W.S. Cell cycle control as a basis for cancer drug development. Int. J. Oncol. 2000, 16, 871–957. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. development. Cdk inhibitors: Positive and negative regulators of g1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Rathinam, M.K.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. Jove (J. Vis. Exp.) 2014, e51998. [Google Scholar] [CrossRef]
- Bolte, C.; Flood, H.M.; Ren, X.; Jagannathan, S.; Barski, A.; Kalin, T.V.; Kalinichenko, V.V. Foxf1 transcription factor promotes lung regeneration after partial pneumonectomy. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, P.; Li, W.; Yang, Y.; Tian, Y.; Wang, X.; Chen, S.; Yang, Y.; Huang, T.; Zhao, T. Kdm1a promotes tumor cell invasion by silencing timp3 in non-small cell lung cancer cells. Oncotarget 2016, 7, 27959. [Google Scholar] [CrossRef]
- Lo, P.H.Y.; Lung, H.L.; Cheung, A.K.L.; Apte, S.S.; Chan, K.W.; Kwong, F.M.; Ko, J.M.Y.; Cheng, Y.; Law, S.; Srivastava, G. Extracellular protease adamts9 suppresses esophageal and nasopharyngeal carcinoma tumor formation by inhibiting angiogenesis. Cancer Res. 2010, 70, 5567–5576. [Google Scholar] [CrossRef]
- Du, W.; Wang, S.; Zhou, Q.; Li, X.; Chu, J.; Chang, Z.; Tao, Q.; Ng, E.; Fang, J.; Sung, J.; et al. Adamts9 is a functional tumor suppressor through inhibiting akt/mtor pathway and associated with poor survival in gastric cancer. Oncogene 2013, 32, 3319–3328. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, Z.; Deng, Z.; Zhou, Y.; Gong, Q.; Zhao, R.; Chen, T. Upregulated lncrna adamts9-as2 suppresses progression of lung cancer through inhibition of mir-223-3p and promotion of tgfbr3. Iubmb Life 2018, 70, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. Trrust v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Sasaki, Y.; Koyama, R.; Takeda, K.; Idogawa, M.; Tokino, T. Forkhead transcription factor foxf1 is a novel target gene of the p53 family and regulates cancer cell migration and invasiveness. Oncogene 2014, 33, 4837–4846. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and ig-cams in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N. Tumor suppressor gene e-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef]
- Berx, G.; Cleton-Jansen, A.; Nollet, F.; De Leeuw, W.; Van de Vijver, M.; Cornelisse, C.; Van Roy, F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995, 14, 6107–6115. [Google Scholar] [CrossRef]
- Bussemakers, M.J.; Vanbokhoven, A.; Voller, M.; Smit, F.P.; Schalken, J.A. The genes for the calcium-dependent cell adhesion molecules p-and e-cadherin are tandemly arranged in the human genome. Biochem. Biophys. Res. Commun. 1994, 203, 1291–1294. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-Y.; Chan, C.-H.; Dubey, N.K.; Wei, H.-J.; Lu, J.-H.; Chang, C.-C.; Cheng, H.-C.; Ou, K.-L.; Deng, W.-P. Highly Expressed FOXF1 Inhibit Non-Small-Cell Lung Cancer Growth via Inducing Tumor Suppressor and G1-Phase Cell-Cycle Arrest. Int. J. Mol. Sci. 2020, 21, 3227. https://doi.org/10.3390/ijms21093227
Wu C-Y, Chan C-H, Dubey NK, Wei H-J, Lu J-H, Chang C-C, Cheng H-C, Ou K-L, Deng W-P. Highly Expressed FOXF1 Inhibit Non-Small-Cell Lung Cancer Growth via Inducing Tumor Suppressor and G1-Phase Cell-Cycle Arrest. International Journal of Molecular Sciences. 2020; 21(9):3227. https://doi.org/10.3390/ijms21093227
Chicago/Turabian StyleWu, Chia-Yu, Chun-Hao Chan, Navneet Kumar Dubey, Hong-Jian Wei, Jui-Hua Lu, Chun-Chao Chang, Hsin-Chung Cheng, Keng-Liang Ou, and Win-Ping Deng. 2020. "Highly Expressed FOXF1 Inhibit Non-Small-Cell Lung Cancer Growth via Inducing Tumor Suppressor and G1-Phase Cell-Cycle Arrest" International Journal of Molecular Sciences 21, no. 9: 3227. https://doi.org/10.3390/ijms21093227
APA StyleWu, C.-Y., Chan, C.-H., Dubey, N. K., Wei, H.-J., Lu, J.-H., Chang, C.-C., Cheng, H.-C., Ou, K.-L., & Deng, W.-P. (2020). Highly Expressed FOXF1 Inhibit Non-Small-Cell Lung Cancer Growth via Inducing Tumor Suppressor and G1-Phase Cell-Cycle Arrest. International Journal of Molecular Sciences, 21(9), 3227. https://doi.org/10.3390/ijms21093227