SARS Unique Domain (SUD) of Severe Acute Respiratory Syndrome Coronavirus Induces NLRP3 Inflammasome-Dependent CXCL10-Mediated Pulmonary Inflammation


 ,
, 
Abstract
1. Introduction
2. Results
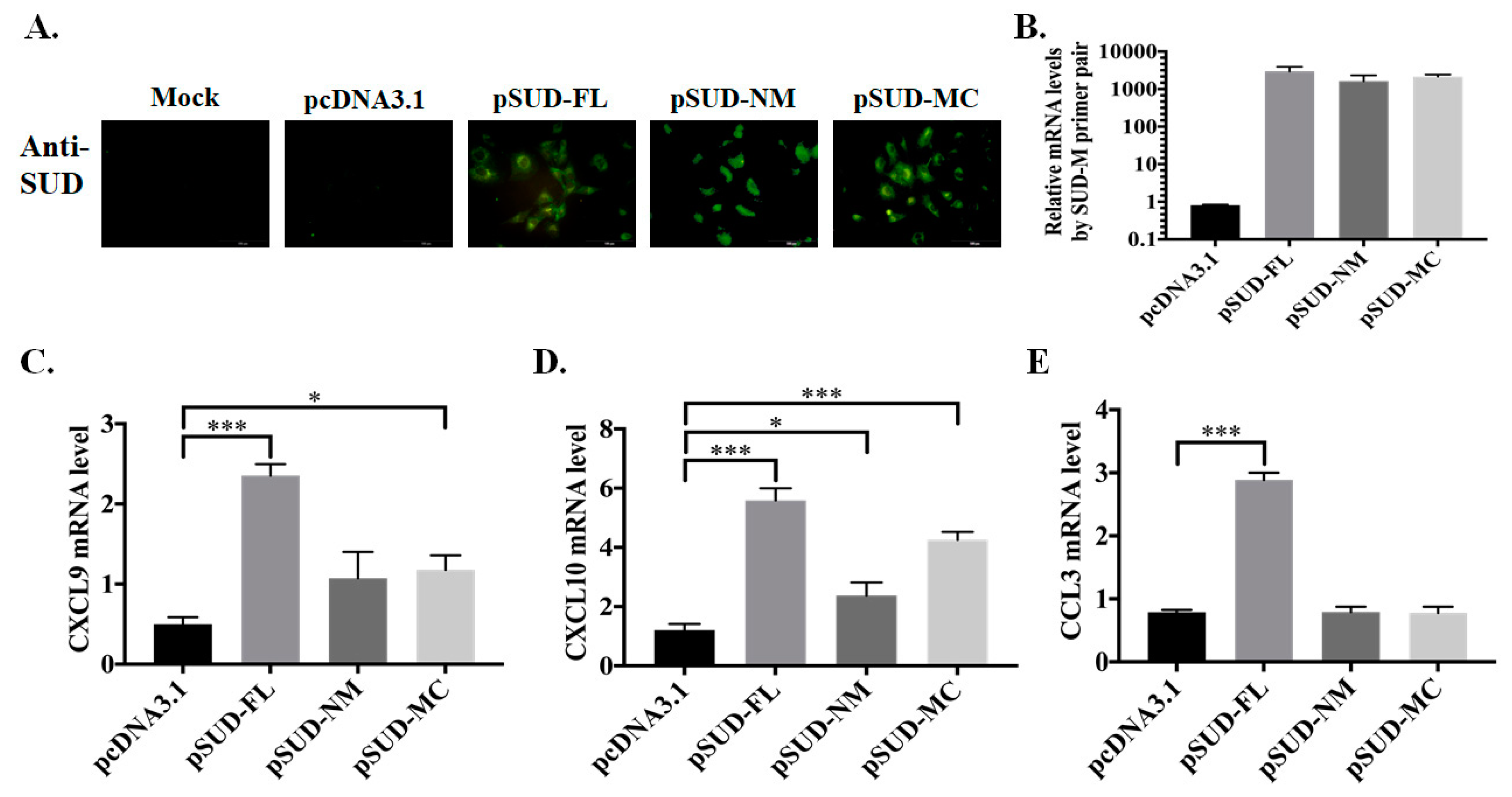
2.1. SUD-MC Subdomain Up-Regulated the Expression of CXCL10 in Human Lung Epithelial Cells
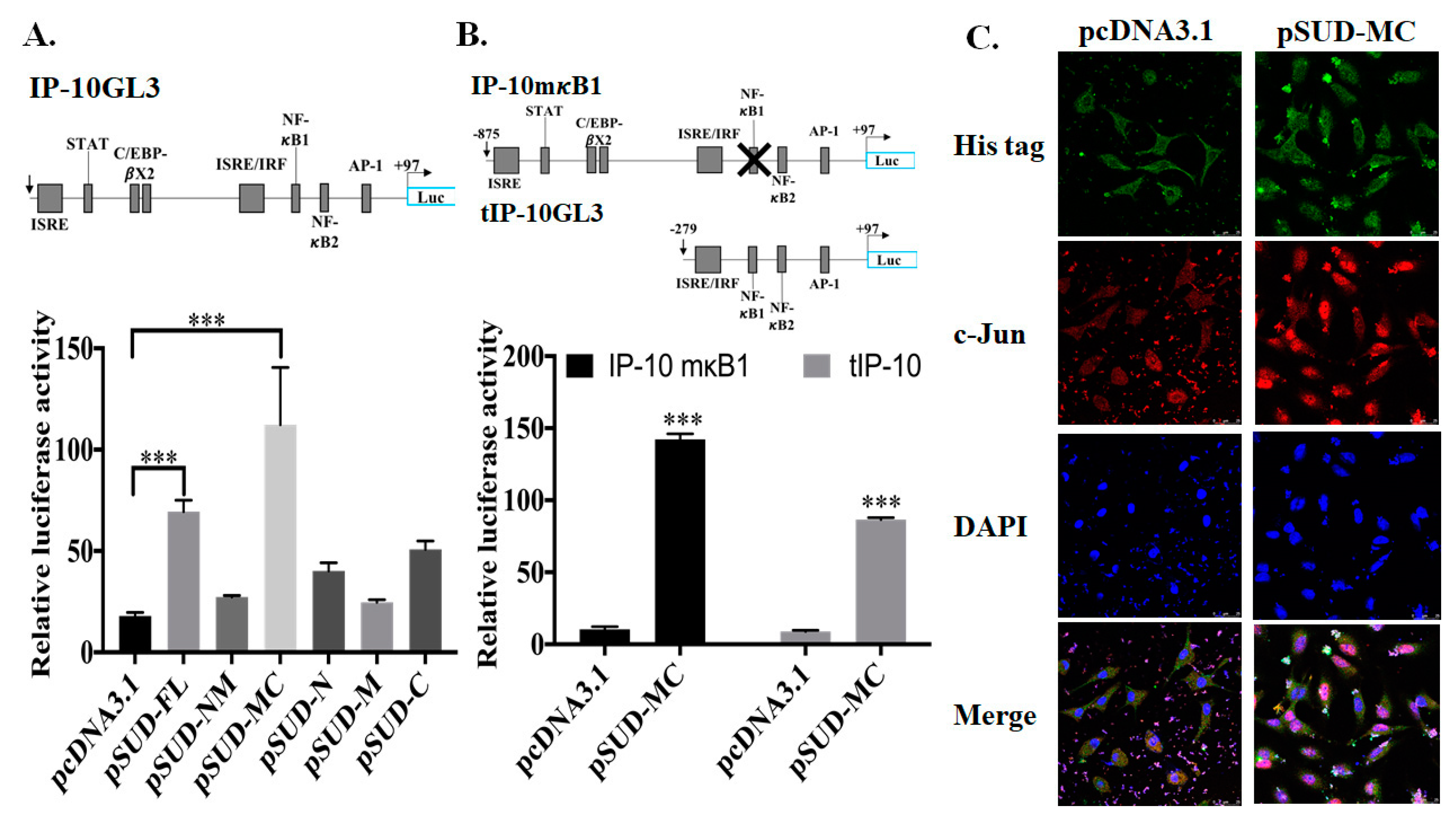
2.2. SUD-MC Subdomain Activated AP1-Mediated Activation of the CXCL10 Promoter
2.3. SUD-MC Significantly Induced the Pulmonary Infiltration of Immune Cells and Caused Lung Injury in Mice
2.4. SARS-CoV SUD-MC Activated NLRP3 Inflammasome-Mediated Up-Regulation of CXCL10 in Pulmonary Inflammation
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Plasmids
4.3. Transient Transfection and Stable Clone Cell Line Generation
4.4. Real-Time RT-PCR
4.5. Dual-Luciferase Reporter Assay
4.6. Immunofluorescence Staining
4.7. Direct Enzyme-Linked Immunosorbent Assay
4.8. Western Blot and Dot Blot Assays
4.9. Animal Model
4.10. Flow Cytometry Assay
4.11. Histopathology and Immunohistochemistry Assays
4.12. Inhibitor Treatment
4.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.; Dong, X.P.; Jiang, G.; Peiris, M. SARS: Clinical virology and pathogenesis. Respirol. 2003, 8, S6–S8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Chang, S.C. Severe acute respiratory syndrome. Curr. Opin. Infect. Dis. 2004, 17, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R.; Peiris, M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013, 100, 286–295. [Google Scholar] [CrossRef]
- Stadler, K.; Masignani, V.; Eickmann, M.; Becker, S.; Abrignani, S.; Klenk, H.D.; Rappuoli, R. SARS—Beginning to understand a new virus. Nat. Rev. Microbiol. 2003, 1, 209–218. [Google Scholar] [CrossRef]
- Min, C.K.; Cheon, S.; Ha, N.Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.Y.; Inn, K.S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef]
- Gillim-Ross, L.; Taylor, J.; Scholl, D.R.; Ridenour, J.; Masters, P.S.; Wentworth, D.E. Discovery of novel human and animal cells infected by the severe acute respiratory syndrome coronavirus by replication-specific multiplex reverse transcription-PCR. J. Clin. Microbiol. 2004, 42, 3196–3206. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- He, L.; Ding, Y.; Zhang, Q.; Che, X.; He, Y.; Shen, H.; Wang, H.; Li, Z.; Zhao, L.; Geng, J.; et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 2006, 210, 288–297. [Google Scholar] [CrossRef]
- Cameron, M.J.; Ran, L.; Xu, L.; Danesh, A.; Bermejo-Martin, J.F.; Cameron, C.M.; Muller, M.P.; Gold, W.L.; Richardson, S.E.; Poutanen, S.M.; et al. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 2007, 81, 8692–8706. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Choe, P.G.; Park, W.B.; Oh, H.S.; Kim, E.J.; Nam, E.Y.; Na, S.H.; Kim, M.; Song, K.H.; Bang, J.H.; et al. Clinical Progression and Cytokine Profiles of Middle East Respiratory Syndrome Coronavirus Infection. J. Korean Med. Sci. 2016, 31, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus. Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Cotten, M.; Watson, S.J.; Kellam, P.; Al-Rabeeah, A.A.; Makhdoom, H.Q.; Assiri, A.; Al-Tawfiq, J.A.; Alhakeem, R.F.; Madani, H.; AlRabiah, F.A.; et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive genomic study. Lancet 2013, 382, 1993–2002. [Google Scholar] [CrossRef]
- Neuman, B.W.; Joseph, J.S.; Saikatendu, K.S.; Serrano, P.; Chatterjee, A.; Johnson, M.A.; Liao, L.; Klaus, J.P.; Yates, J.R., 3rd; Wuthrich, K.; et al. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 2008, 82, 5279–5294. [Google Scholar] [CrossRef]
- Tan, J.; Vonrhein, C.; Smart, O.S.; Bricogne, G.; Bollati, M.; Kusov, Y.; Hansen, G.; Mesters, J.R.; Schmidt, C.L.; Hilgenfeld, R. The SARS-unique domain (SUD) of SARS coronavirus contains two macrodomains that bind G-quadruplexes. PLoS Pathog. 2009, 5, e1000428. [Google Scholar] [CrossRef]
- Kusov, Y.; Tan, J.; Alvarez, E.; Enjuanes, L.; Hilgenfeld, R. A G-quadruplex-binding macrodomain within the “SARS-unique domain” is essential for the activity of the SARS-coronavirus replication-transcription complex. Virology 2015, 484, 313–322. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Muller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Tan, J.; Kusov, Y.; Mutschall, D.; Tech, S.; Nagarajan, K.; Hilgenfeld, R.; Schmidt, C.L. The “SARS-unique domain” (SUD) of SARS coronavirus is an oligo(G)-binding protein. Biochem. Biophys. Res. Commun. 2007, 364, 877–882. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Goldmann, T.; Ludwig, C.; Prasse, A.; Vollmer, E.; Muller-Quernheim, J.; Zissel, G. CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir. Res. 2005, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Liu, S.; Pan, P.; Su, X.; Tan, H.; Wu, D.; Zhang, L.; Song, C.; Dai, M.; et al. Pirfenidone ameliorates lipopolysaccharide-induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol. Immunol. 2018, 99, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, J.; Zhou, C.; Wu, Z.; Zhong, S.; Liu, J.; Luo, W.; Chen, T.; Qin, Q.; Deng, P. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care. Med. 2005, 171, 850–857. [Google Scholar] [CrossRef]
- Spiegel, M.; Weber, F. Inhibition of cytokine gene expression and induction of chemokine genes in non-lymphatic cells infected with SARS coronavirus. Virol. J. 2006, 3, 17. [Google Scholar] [CrossRef]
- Yen, Y.T.; Liao, F.; Hsiao, C.H.; Kao, C.L.; Chen, Y.C.; Wu-Hsieh, B.A. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J. Virol. 2006, 80, 2684–2693. [Google Scholar] [CrossRef]
- Glass, W.G.; Subbarao, K.; Murphy, B.; Murphy, P.M. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J. Immunol. 2004, 173, 4030–4039. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schafer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. M. Bio. 2015, 6, e00615–e00638. [Google Scholar] [CrossRef]
- Roberts, A.; Deming, D.; Paddock, C.D.; Cheng, A.; Yount, B.; Vogel, L.; Herman, B.D.; Sheahan, T.; Heise, M.; Genrich, G.L.; et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007, 3, e5. [Google Scholar] [CrossRef]
- Wentworth, D.E.; Gillim-Ross, L.; Espina, N.; Bernard, K.A. Mice susceptible to SARS coronavirus. Emerg. Infect. Dis. 2004, 10, 1293–1296. [Google Scholar] [CrossRef]
- Law, A.H.; Lee, D.C.; Cheung, B.K.; Yim, H.C.; Lau, A.S. Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J. Virol. 2007, 81, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Holzberg, D.; Knight, C.G.; Dittrich-Breiholz, O.; Schneider, H.; Dorrie, A.; Hoffmann, E.; Resch, K.; Kracht, M. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN delta domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J. Biol. Chem. 2003, 278, 40213–40223. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Haronikova, L.; Liao, J.C.; Fojta, M. DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Mou, Y.; Bernstock, J.D.; Klimanis, D.; Wang, S.; Spatz, M.; Maric, D.; Johnson, K.; Klinman, D.M.; Li, X.; et al. Synthetic Oligodeoxynucleotides Containing Multiple Telemeric TTAGGG Motifs Suppress Inflammasome Activity in Macrophages Subjected to Oxygen and Glucose Deprivation and Reduce Ischemic Brain Injury in Stroke-Prone Spontaneously Hypertensive Rats. PLoS ONE 2015, 10, e0140772. [Google Scholar] [CrossRef]
- Ahn, M.; Anderson, D.E.; Zhang, Q.; Tan, C.W.; Lim, B.L.; Luko, K.; Wen, M.; Chia, W.N.; Mani, S.; Wang, L.C.; et al. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat. Microbiol. 2019, 4, 789–799. [Google Scholar] [CrossRef]
- Wang, J.M.; Wang, L.F.; Shi, Z.L. Construction of a non-infectious SARS coronavirus replicon for application in drug screening and analysis of viral protein function. Biochem. Biophys. Res. Commun. 2008, 374, 138–142. [Google Scholar] [CrossRef]
- Li, S.W.; Wang, C.Y.; Jou, Y.J.; Yang, T.C.; Huang, S.H.; Wan, L.; Lin, Y.J.; Lin, C.W. SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci. Rep. 2016, 6, 25754. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Fragments | Primer Name | Primer Sequence |
|---|---|---|
| full-length SUD | F-SUD-N | 5′-AGTG GGTACC T ATTAAGGCCTGCATTGATGAG-3′ |
| R-SUD-C | 5′-AGTG CTCGAG CG TGTGTGGAGATTAGTGTTGTC-3′ | |
| SUD-NM | F-SUD-N | 5′-AGTG GGTACC T ATTAAGGCCTGCATTGATGAG-3′ |
| R-SUD-M | 5′-AGTG CTCGAG CG TGACGAAGTGAGGTATCC-3′ | |
| SUD-MC | F-SUD-M | 5′-AGTG GGTACC T TGGAATTTGAGAGAAATG-3′ |
| R-SUD-C | 5′-AGTG CTCGAG CG TGTGTGGAGATTAGTGTTGTC-3′ | |
| SUD-N | F-SUD-N | 5′-AGTG GGTACC T ATTAAGGCCTGCATTGATGAG-3′ |
| R-SUD-N | 5′-AGTG CTCGAG CG TAGAATCTCTTCCTTAGC-3′ | |
| SUD-M | F-SUD-M | 5′-AGTG GGTACC T TGGAATTTGAGAGAAATG-3′ |
| R-SUD-M | 5′-AGTG CTCGAG CG TGACGAAGTGAGGTATCC-3′ | |
| SUD-C | F-SUD-C | 5′-AGTG GGTACC T TCAAAGACATCTGAGGAG-3′ |
| R-SUD-C | 5′-AGTG CTCGAG CG TGTGTGGAGATTAGTGTTGTC-3′ |
| Species | Gene | Forward Primer Sequences | Reverse Primer Sequences |
|---|---|---|---|
| SARS-CoV | SUD-N | TCAGAACATGCTTAGAGG | TGGAGGGTATTACAACACAA |
| SUD-M | CATGCTGAAGAGACAAGAAAAT | AGTATAAAAGAAGAATCGGACACC | |
| Human | IL-1β | ATCACTGAACTGCACGCTCC | TTGTTCTCCATATCCTGTCCC |
| CXCL8 | CGATGTCAGTGGATAAAGACA | TGAATTCTCAGCCCTCTTCAAAAA | |
| CXCL9 | CGTGGTAAAACACTTGCGGATATT | CAATCATGCTTCCACTAACCGACT | |
| CXCL10 | CCAATTTTGTCCACGTGTTG | TTCTTGATGGCCTTCGATTC | |
| CCL3 | AGCTGACTACTTTGAGACGAGCA | CGGCTTCGCTTGGTTAGGA | |
| CCL5 | TCCCCATATTCCTCGGAC | GTCTAGAGGAACCGGTGTTAC | |
| β-actin | AGGCCACCCCAGAGGACAAC | CCAGAGGCGTACAGGGATA | |
| Mouse | IL-1β | CCAGCAGGTTATCATCATCATCC | CTCGCAGCAGCACATCAAC |
| CXCL9 | GCCATGAAGTCCGCTGTTCT | GGGTTCCTCGAACTCCACACT | |
| CXCL10 | GACGGTCCGCTGCAACTG | GCTTCCCTATGGCCCTCATT | |
| CCL3 | TGAAACCAGCAGCCTTTGCTC | AGGCATTCAGTTCCAGGTCAGTG | |
| CCL5 | GATGGACATAGAGGACACAACT | TGGGACGGCAGATCTGAGGG | |
| GAPDH | TGAGGCCGGTGCTGAGTATGTCG | CCACAGTCTTCTGGGTGGCAGTG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-S.; Ko, B.-H.; Ju, J.-C.; Chang, H.-H.; Huang, S.-H.; Lin, C.-W. SARS Unique Domain (SUD) of Severe Acute Respiratory Syndrome Coronavirus Induces NLRP3 Inflammasome-Dependent CXCL10-Mediated Pulmonary Inflammation. Int. J. Mol. Sci. 2020, 21, 3179. https://doi.org/10.3390/ijms21093179
Chang Y-S, Ko B-H, Ju J-C, Chang H-H, Huang S-H, Lin C-W. SARS Unique Domain (SUD) of Severe Acute Respiratory Syndrome Coronavirus Induces NLRP3 Inflammasome-Dependent CXCL10-Mediated Pulmonary Inflammation. International Journal of Molecular Sciences. 2020; 21(9):3179. https://doi.org/10.3390/ijms21093179
Chicago/Turabian StyleChang, Young-Sheng, Bo-Han Ko, Jyh-Cherng Ju, Hsin-Hou Chang, Su-Hua Huang, and Cheng-Wen Lin. 2020. "SARS Unique Domain (SUD) of Severe Acute Respiratory Syndrome Coronavirus Induces NLRP3 Inflammasome-Dependent CXCL10-Mediated Pulmonary Inflammation" International Journal of Molecular Sciences 21, no. 9: 3179. https://doi.org/10.3390/ijms21093179
APA StyleChang, Y.-S., Ko, B.-H., Ju, J.-C., Chang, H.-H., Huang, S.-H., & Lin, C.-W. (2020). SARS Unique Domain (SUD) of Severe Acute Respiratory Syndrome Coronavirus Induces NLRP3 Inflammasome-Dependent CXCL10-Mediated Pulmonary Inflammation. International Journal of Molecular Sciences, 21(9), 3179. https://doi.org/10.3390/ijms21093179