Nifedipine Upregulates ATF6-α, Caspases -12, -3, and -7 Implicating Lipotoxicity-Associated Renal ER Stress

 , and
, and
Abstract
1. Introduction
2. Results
2.1. NF Aggravated Renal Function in Rats with Doxorubicin (DR)-Induced CKD
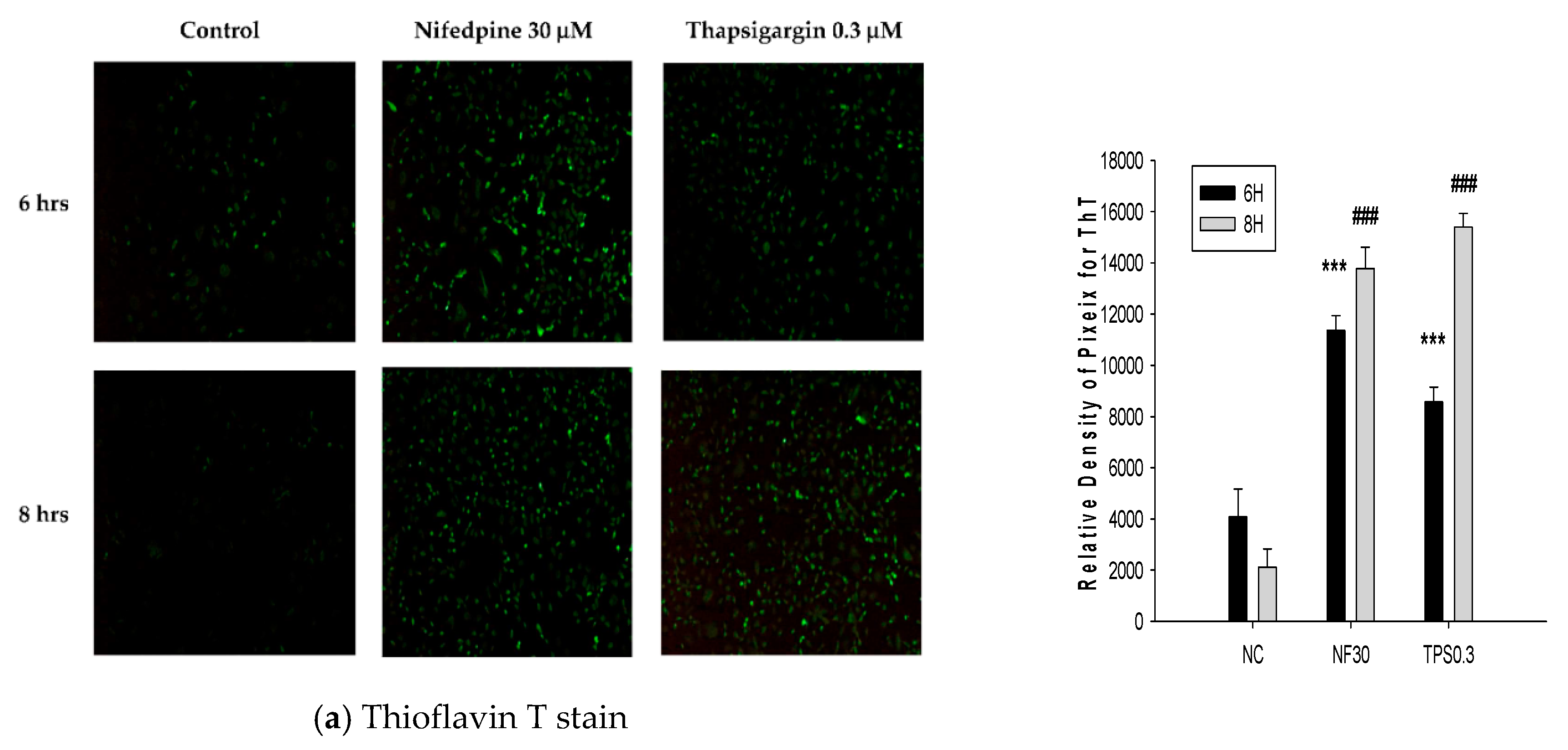
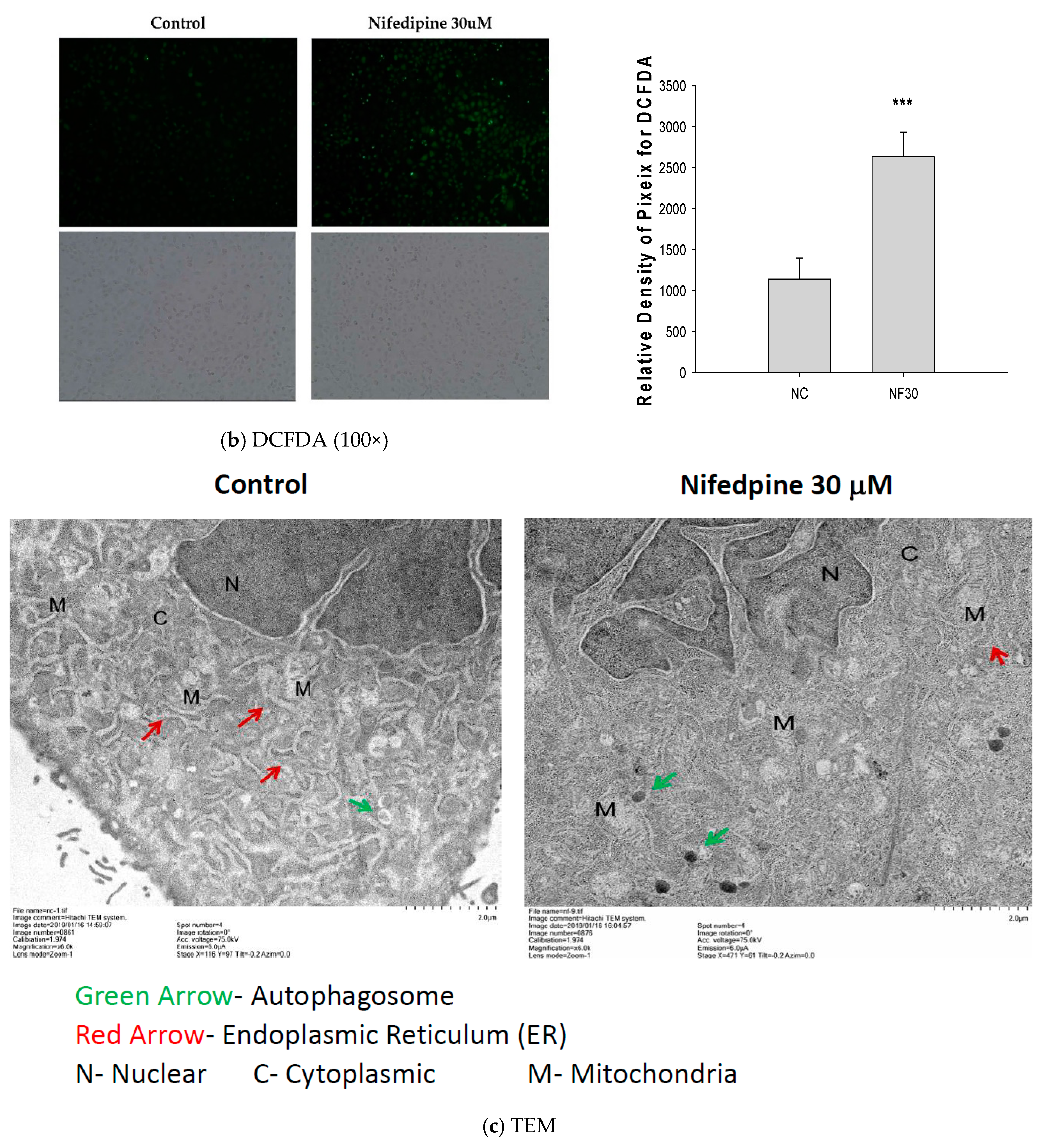
2.2. NF Triggered Accumulation of Misfolded Proteins, Production of Reactive Oxygen Species (ROS), and Disruption of ER Folding
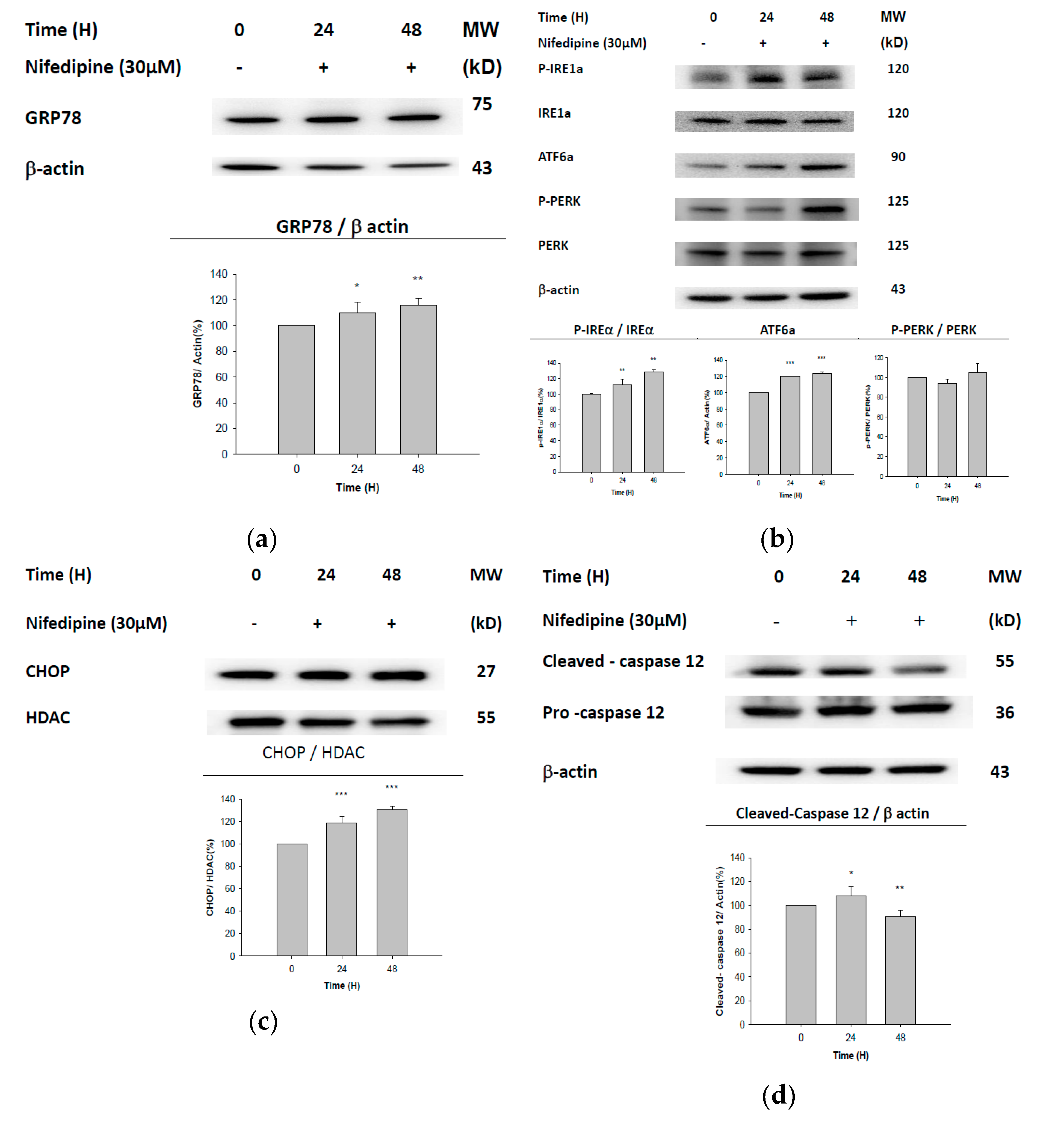
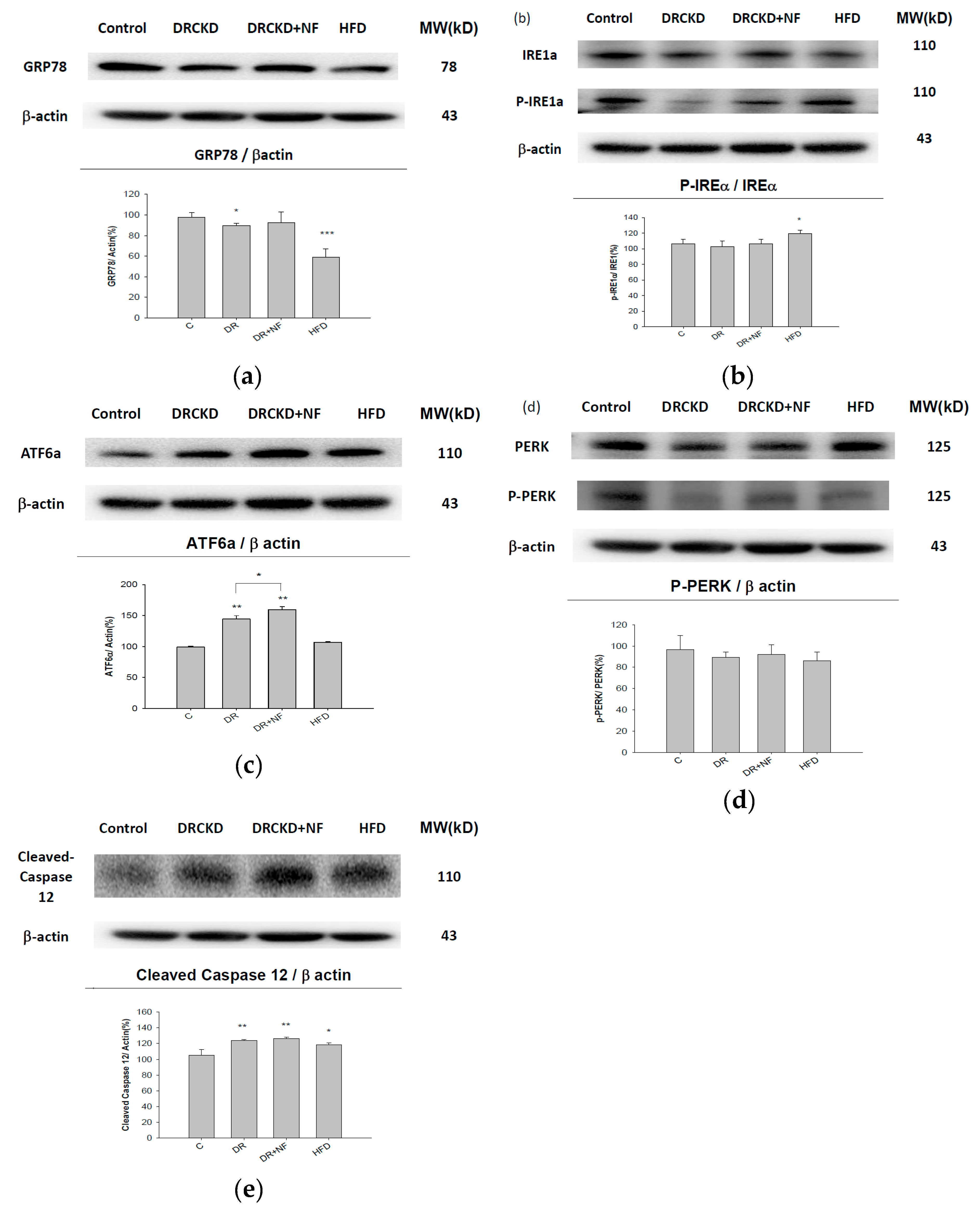
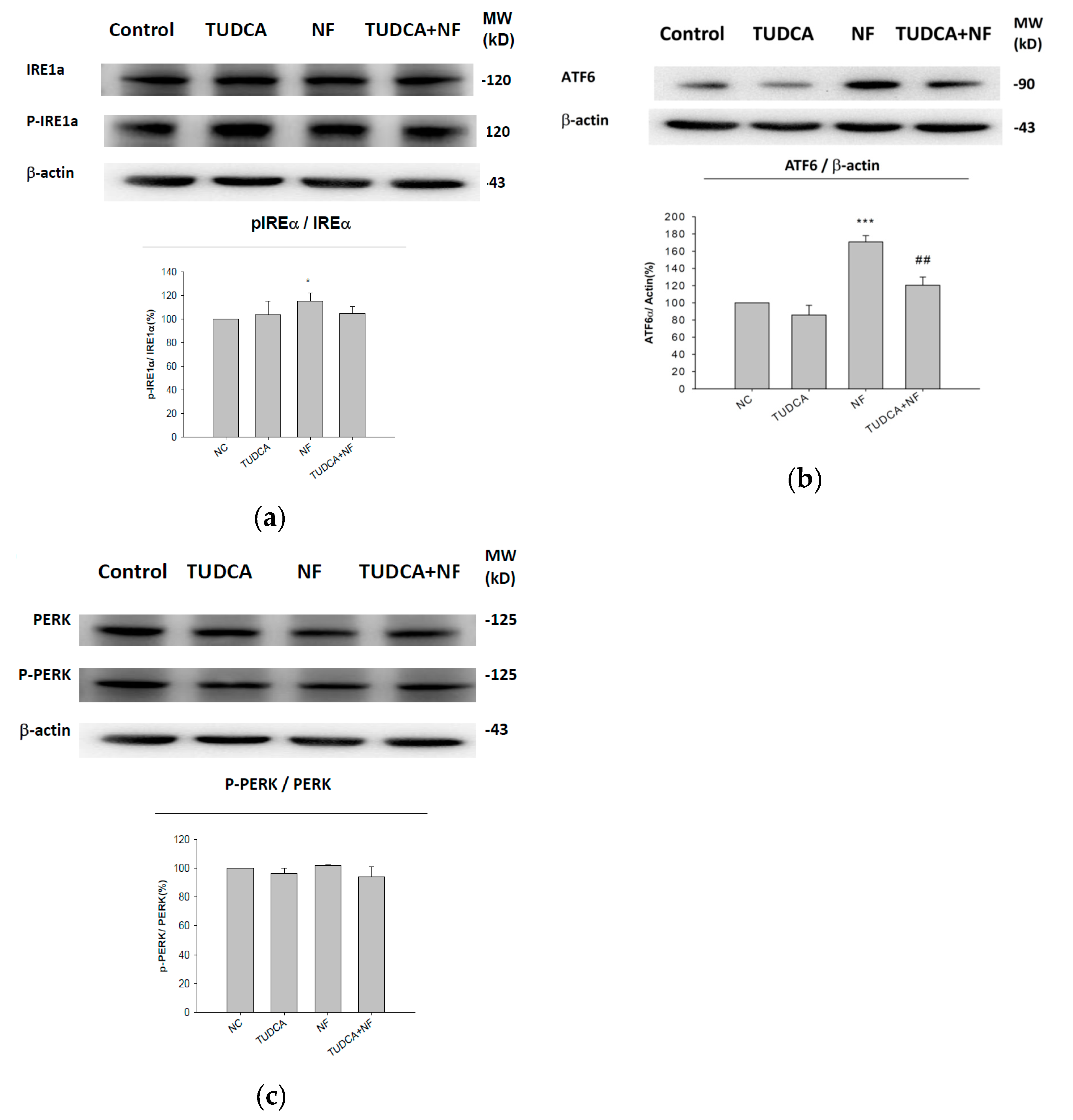
2.3. UPR-ER Stress-Related Protein Signals Were Significantly Affected in NRK52E Cells In Vitro and Kidney Tissues In Vivo
2.4. ATF6α Was Confirmed to Be the Only ER Stress Pathway Affected by NF
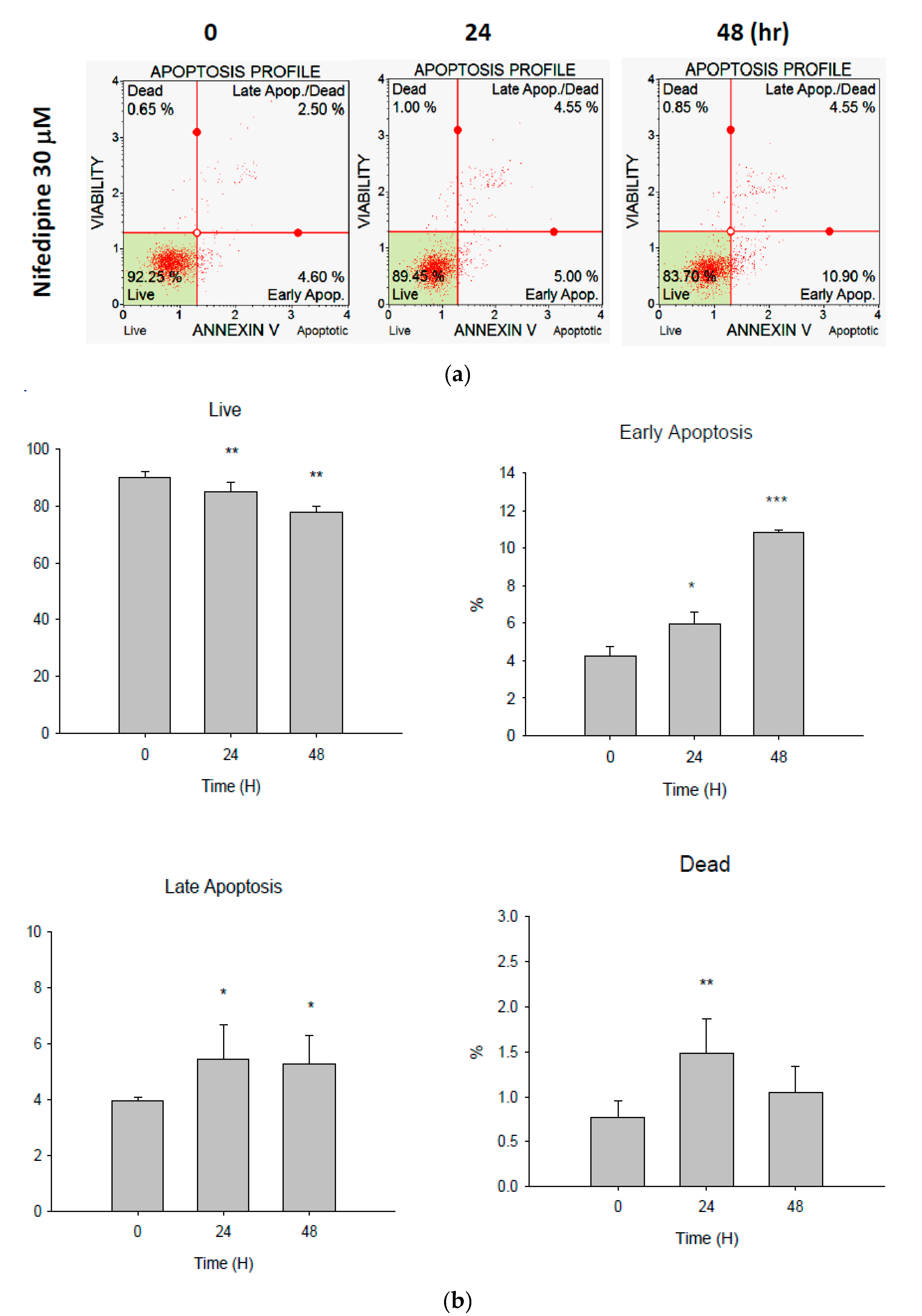
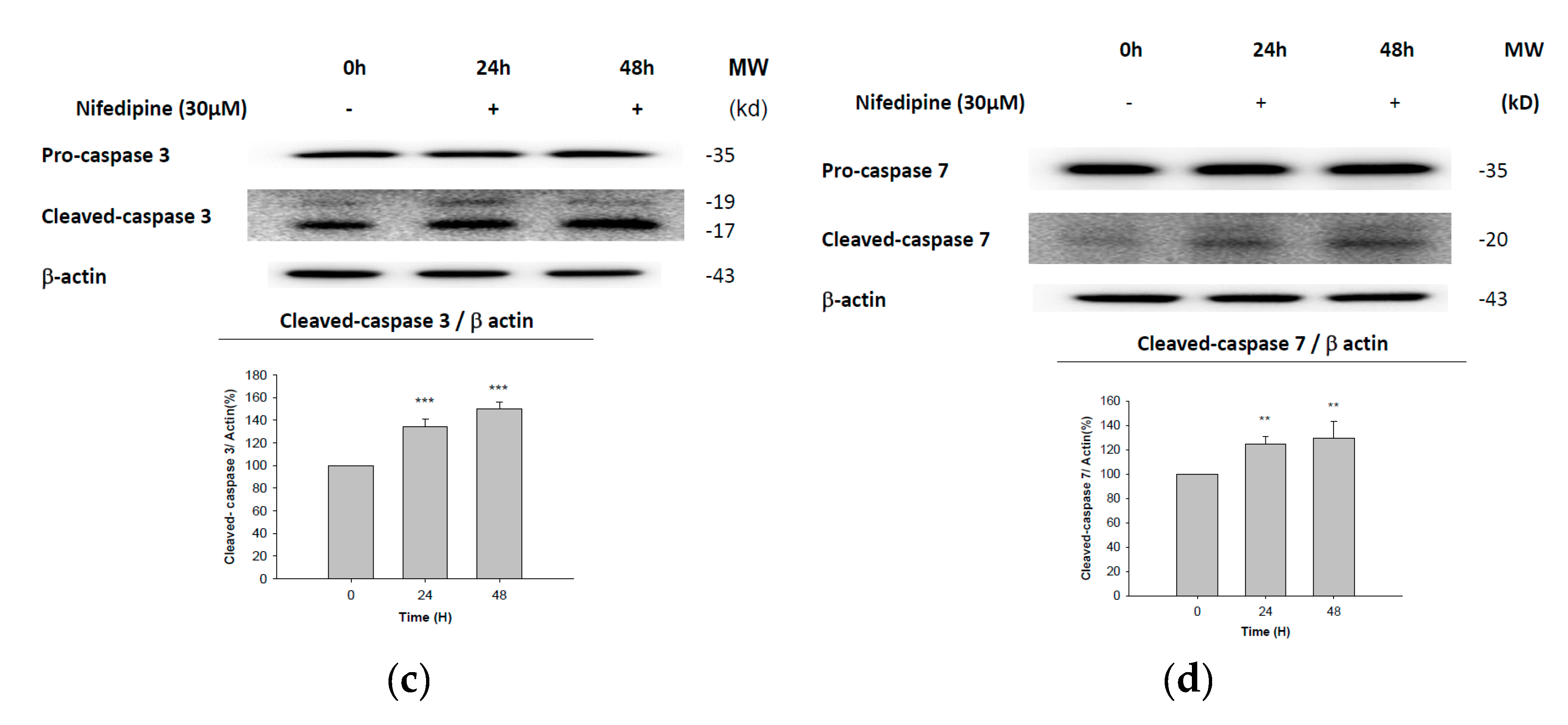
2.5. Early Apoptosis of NRK52E Cells Induced by NF May Partially Be Due to Activation of Caspase-3 and Caspase-7
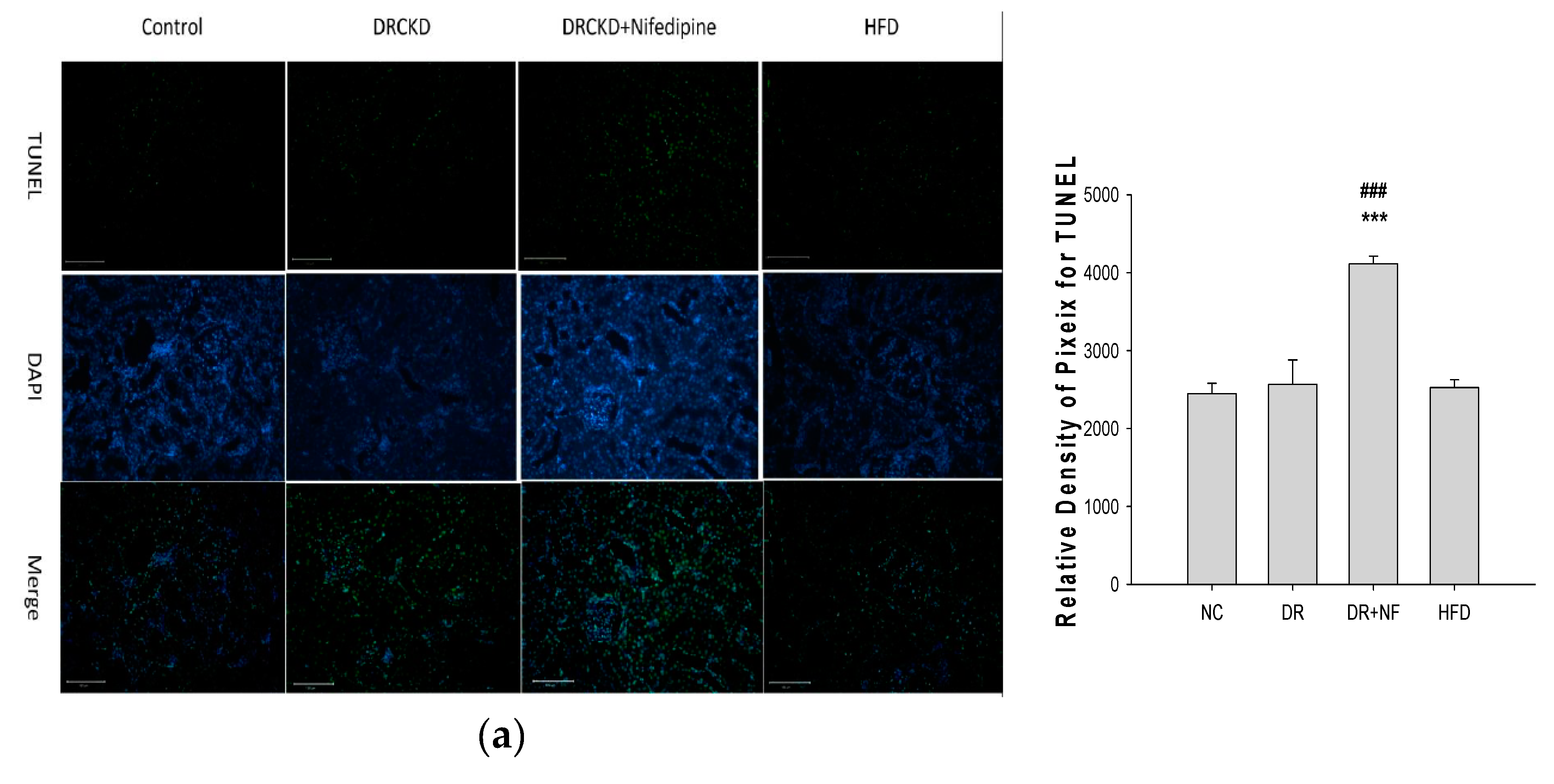
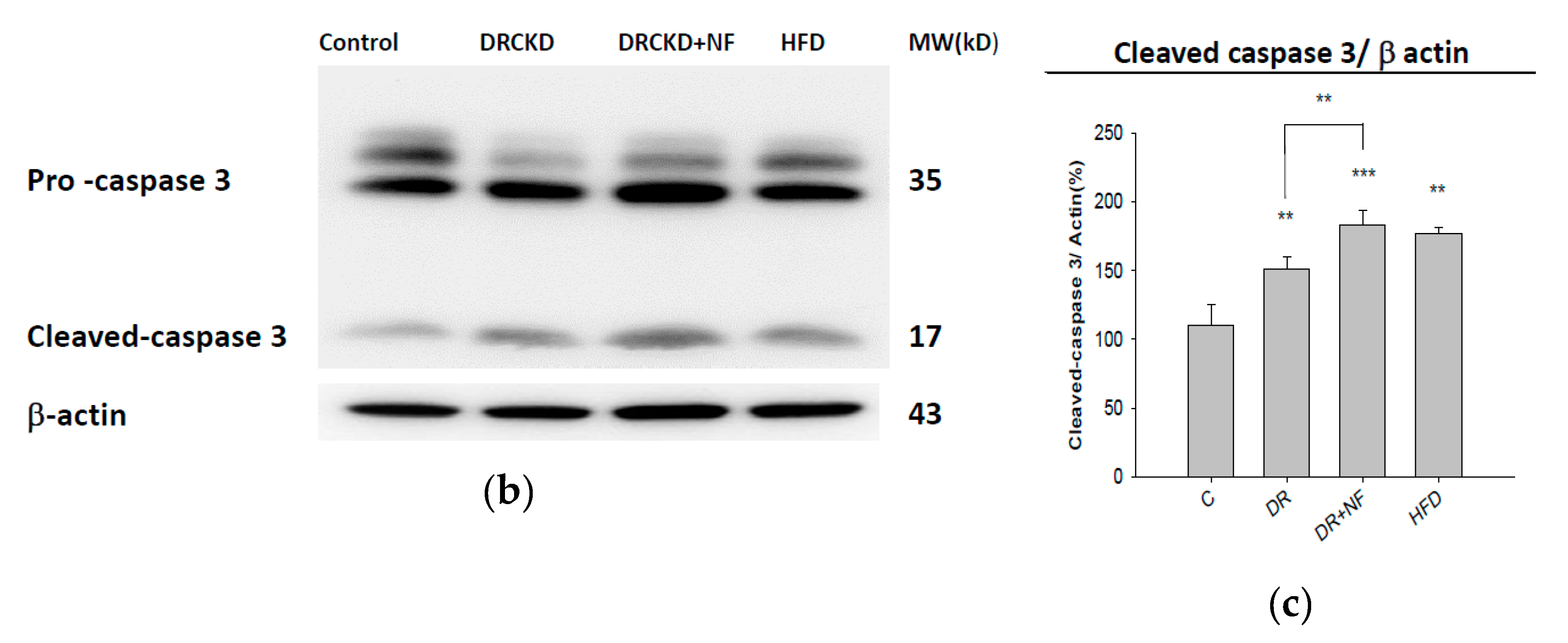
2.6. NF Also Enhanced Renal Apoptosis and Expression of Cleaved Caspase-3 in Rats with Experimentally Induced CKD
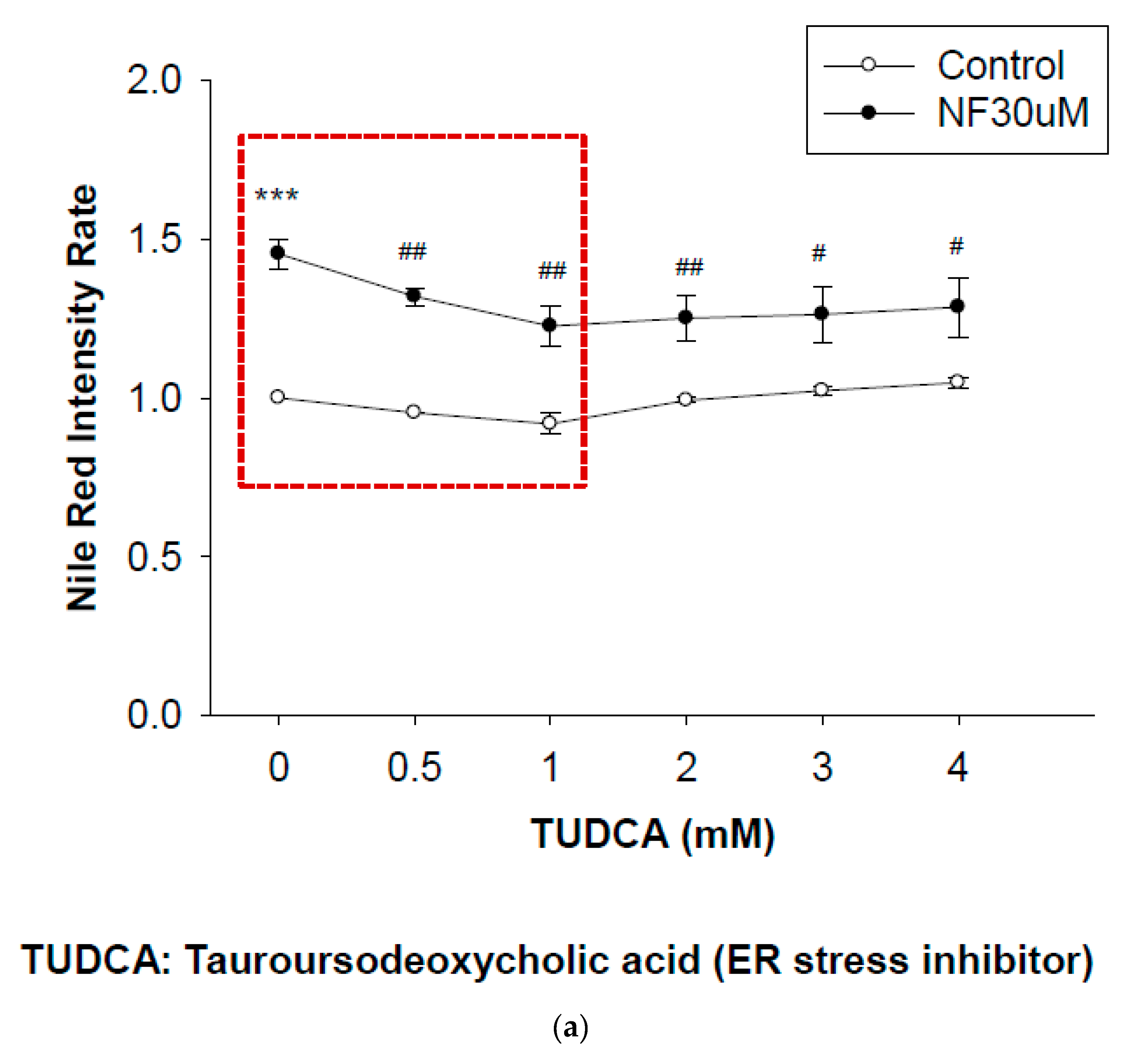
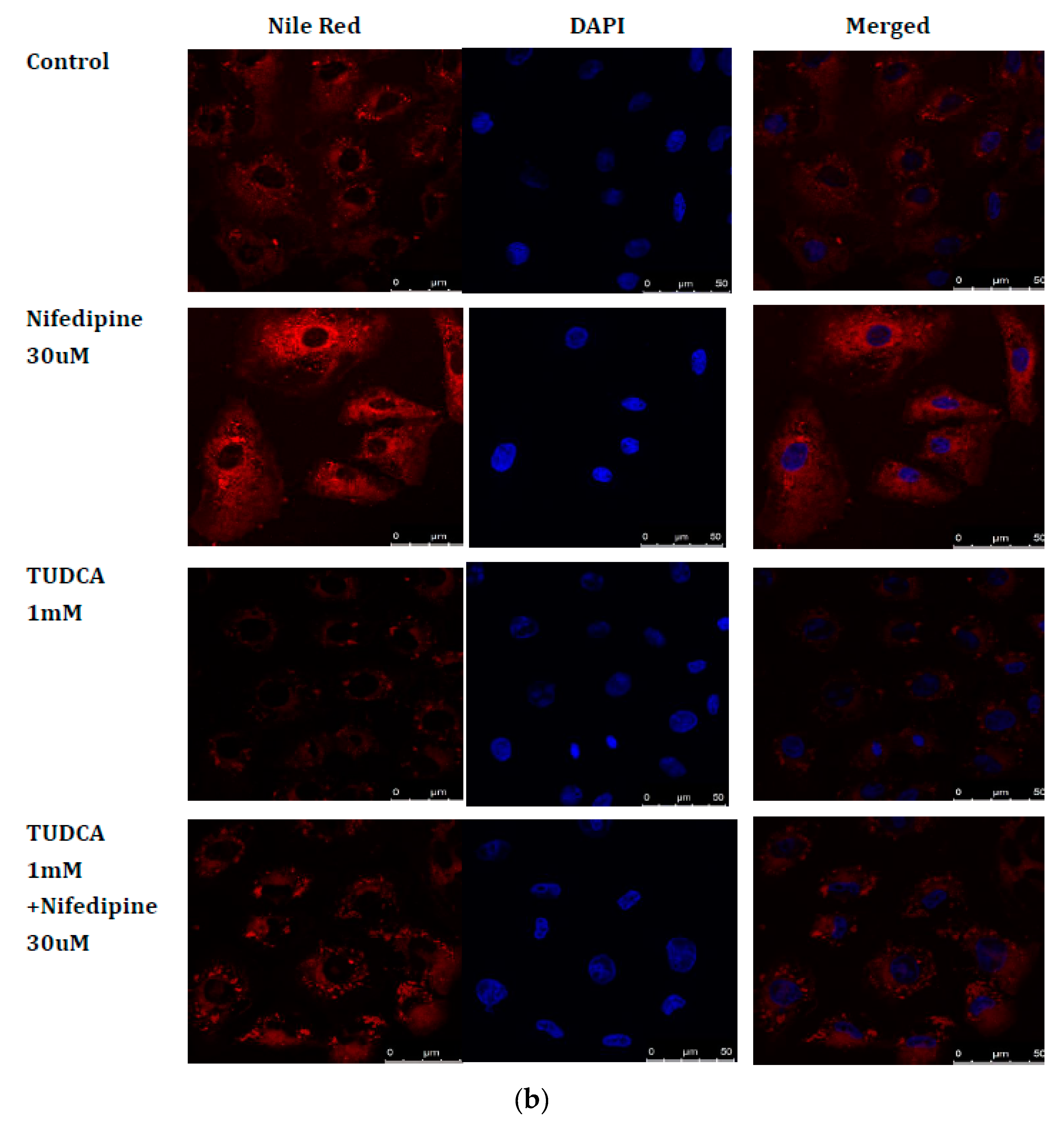
2.7. Blockade of ER Stress Ameliorates Lipid Accumulation Induced by NF
3. Discussion
4. Materials and Methods
4.1. Chemicals and Kits
4.2. Cell Culture
4.3. Animal Experiments
4.4. Western Blotting
4.5. Thioflavin T (ThT) Stain
4.6. 2′,7′-Dichlorofluorescin Diacetate (DCFDA) Staining
4.7. Transmission Electron Microscopy (TEM)
4.8. Terminal Deoxynucleotidyl Transferase (TdT) dUTP Nick End Labeling (TUNEL) Assay
4.9. Nile Red Confocal Staining
4.10. Annexin V/Propidium Iodide (PI) Flow Cytometry
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenyl butyric acid |
| ATF6 | activating transcription factor 6 |
| CHOP | C/EBP homologous protein |
| CKD | chronic kidney disease |
| CsA | cyclosporine A |
| DCFDA | 2′,7′-dichlorofluorescin diacetate |
| DR | doxorubicin |
| EMT | epithelial-to-mesenchymal transition |
| ER | endoplasmic reticulum |
| ERSE | ER stress element |
| HFD | high-fat diet |
| IRE1 | inositol-requiring enzyme 1 |
| NF | nifedipine |
| PBS | phosphate-buffered saline |
| PERK | PKR-like ER kinase |
| SREBP | sterol regulatory element-binding protein |
| TEM | transmission electron microscopy |
| ThT | thioflavin T |
| TPS | thapsigargin |
| TUDCA | tauroursodeoxycholic acid |
| TUNEL | terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling |
| UPR | unfolded protein response |
References
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Austin, R.C. ER stress and lipogenesis: A slippery slope toward hepatic steatosis. Dev. Cell 2008, 15, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Jerry Chiang, W.C.; Lin, J.H. The effects of IRE1, ATF6, and PERK signaling on adRP-linked rhodopsins. Adv. Exp. Med. Biol. 2014, 801, 661–667. [Google Scholar]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Mohammed-Ali, Z.; Lu, C.; Marway, M.K.; Carlisle, R.E.; Ask, K.; Lukic, D.; Krepinsky, J.C.; Dickhout, J.G. Endoplasmic reticulum stress inhibition attenuates hypertensive chronic kidney disease through reduction in proteinuria. Sci. Rep. 2017, 7, 41572. [Google Scholar] [CrossRef]
- Bek, M.F.; Bayer, M.; Muller, B.; Greiber, S.; Lang, D.; Schwab, A.; August, C.; Springer, E.; Rohrbach, R.; Huber, T.B.; et al. Expression and function of C/EBP homology protein (GADD153) in podocytes. Am. J. Pathol. 2006, 168, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F.; Krebs, C.; Abdulhag, U.N.; Meyer-Schwesinger, C.; Maas, R.; Helmchen, U.; Hilgers, K.F.; Wolf, G.; Stahl, R.A.; Wenzel, U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int. 2008, 73, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Markan, S.; Kohli, H.S.; Joshi, K.; Minz, R.W.; Sud, K.; Ahuja, M.; Anand, S.; Khullar, M. Up regulation of the GRP-78 and GADD-153 and down regulation of Bcl-2 proteins in primary glomerular diseases: A possible involvement of the ER stress pathway in glomerulonephritis. Mol. Cell Biochem. 2009, 324, 131–138. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Rastaldi, M.P.; Ikehata, M.; Neusser, M.A.; Kretzler, M.; Cohen, C.D.; Schlondorff, D. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J. Am. Soc. Nephrol. 2008, 19, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lee, K.; Wang, N.; He, J.C. The Role of Endoplasmic Reticulum Stress in Diabetic Nephropathy. Curr. Diabetes Rep. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 2010, 77, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Hsu, S.P.; Wu, C.T.; Huang, J.W.; Cheng, H.T.; Chang, Y.W.; Hung, K.Y.; Wu, K.D.; Liu, S.H. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol. Med. 2011, 17, 1295–1305. [Google Scholar] [CrossRef]
- Syeda, K.; Mohammed, A.M.; Arora, D.K.; Kowluru, A. Glucotoxic conditions induce endoplasmic reticulum stress to cause caspase 3 mediated lamin B degradation in pancreatic beta-cells: Protection by nifedipine. Biochem. Pharm. 2013, 86, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L.; Li, Y.; Chen, H.; Sun, Z. Nifedipine protects INS-1 beta-cell from high glucose-induced ER stress and apoptosis. Int. J. Mol. Sci. 2011, 12, 7569–7580. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, P.; Wang, T.; Chen, K.; Zhu, W.; Wang, H. Inhibition of Calcium Influx Reduces Dysfunction and Apoptosis in Lipotoxic Pancreatic beta-Cells via Regulation of Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0132411. [Google Scholar]
- Irzyniec, T.; Mall, G.; Greber, D.; Ritz, E. Beneficial effect of nifedipine and moxonidine on glomerulosclerosis in spontaneously hypertensive rats. A micromorphometric study. Am. J. Hypertens. 1992, 5, 437–443. [Google Scholar] [CrossRef]
- Grosskopf, I.; Shaish, A.; Charach, G.; Harats, D.; Kamari, Y. Nifedipine Treatment for Hypertension is Associated with Enhanced Lipolytic Activity and Accelerated Clearance of Postprandial Lipemia. Horm. Metab. Res. 2016, 48, 257–262. [Google Scholar] [CrossRef]
- Engineer, F.; Sridhar, R. Inhibition of rat heart and liver microsomal lipid peroxidation by nifedipine. Biochem. Pharm. 1989, 38, 1279–1285. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Wu, M.-S.; Lin, Y.-F.; Chen, C.-R.; Chen, C.-Y.; Chen, C.-J.; Shen, C.-C.; Chen, K.-C.; Peng, C.-C. Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway. Int. J. Mol. Sci. 2019, 20, 1570. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.; Xu, C.; Thibault, G. From the unfolded protein response to metabolic diseases—Lipids under the spotlight. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Katsoulieris, E.; Mabley, J.G.; Samai, M.; Sharpe, M.A.; Green, I.C.; Chatterjee, P.K. Lipotoxicity in renal proximal tubular cells: Relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic. Biol. Med. 2010, 48, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Beriault, D.R.; Werstuck, G.H. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 2293–2301. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar]
- Kass, G.E.; Orrenius, S. Calcium signaling and cytotoxicity. Environ. Health Perspect. 1999, 107, 25–35. [Google Scholar]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. 2005, 4, 14–22. [Google Scholar] [CrossRef]
- Diamond, J.R.; Cheung, J.Y.; Fang, L.S. Nifedipine-induced renal dysfunction. Alterations in renal hemodynamics. Am. J. Med. 1984, 77, 905–909. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Rong, G.; Bu, Y.; Zhang, S.; Zhang, M.; Zhang, J.; Liang, X. Advanced oxidation protein products induce hypertrophy and epithelial-to-mesenchymal transition in human proximal tubular cells through induction of endoplasmic reticulum stress. Cell Physiol. Biochem. 2015, 35, 816–828. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Deng, X.; Xie, Y.; Zhang, A. Advance of autophagy in chronic kidney diseases. Ren Fail 2017, 39, 306–313. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Peng, C.C.; Chen, K.C.; Peng, R.Y. Rutin (quercetin rutinoside) induced protein-energy malnutrition in chronic kidney disease, but quercetin acted beneficially. J. Agric. Food Chem. 2013, 61, 7258–7267. [Google Scholar] [CrossRef]
- Khan, T.J.; Kuerban, A.; Razvi, S.S.; Mehanna, M.G.; Khan, K.A.; Almulaiky, Y.Q.; Faidallah, H.M. In vivo evaluation of hypolipidemic and antioxidative effect of ‘Ajwa’ (Phoenix dactylifera L.) date seed-extract in high-fat diet-induced hyperlipidemic rat model. Biomed. Pharm. 2018, 107, 675–680. [Google Scholar] [CrossRef]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Erguler, K.; Pieri, M.; Deltas, C. A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC Syst. Biol. 2013, 7, 16. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, D.; Ren, J.; Yang, J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim. Biophys. Acta 2015, 1852, 209–218. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Arnold, N.D.; Zechner, D.; Hanford, D.S.; Demartin, K.M.; Mcdonough, P.M.; Prywes, R.; Glembotski, C.C. p38 Mitogen-activated protein kinase mediates the transcriptional induction of the atrial natriuretic factor gene through a serum response element. A potential role for the transcription factor ATF6. J. Biol. Chem. 1998, 273, 20636–20643. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA 1999, 96, 11041–11048. [Google Scholar] [CrossRef]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, Y.; Zeng, C.; He, L.; Wang, N. Tauroursodeoxycholic Acid Attenuates Renal Tubular Injury in a Mouse Model of Type 2 Diabetes. Nutrients 2016, 8. [Google Scholar] [CrossRef]
- Peng, P.; Ma, Q.; Wang, L.; Zhang, O.; Han, H.; Liu, X.; Zhou, Y.; Zhao, Y. Preconditioning With Tauroursodeoxycholic Acid Protects Against Contrast-Induced HK-2 Cell Apoptosis by Inhibiting Endoplasmic Reticulum Stress. Angiology 2015, 66, 941–949. [Google Scholar] [CrossRef]
- Jakubowska, A.; Kilis-Pstrusinska, K. Importance of annexin V in kidney diseases. Postepy Higieny Medycyny Doswiadczalnej (Online) 2015, 69, 153–157. [Google Scholar]
- Sanz, A.B.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Mechanisms of renal apoptosis in health and disease. J. Am. Soc. Nephrol. 2008, 19, 1634–1642. [Google Scholar] [CrossRef]
- Faitova, J.; Krekac, D.; Hrstka, R.; Vojtesek, B. Endoplasmic reticulum stress and apoptosis. Cell Mol. Biol. Lett. 2006, 11, 488–505. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Qiu, M.; Li, S.; Jin, L.; Feng, P.; Kong, Y.; Zhao, X.; Lin, Y.; Xu, Y.; Li, C.; Wang, W. Combination of Chymostatin and Aliskiren attenuates ER stress induced by lipid overload in kidney tubular cells. Lipids Health Dis. 2018, 17, 183. [Google Scholar] [CrossRef]
- Lhotak, S.; Sood, S.; Brimble, E.; Carlisle, R.E.; Colgan, S.M.; Mazzetti, A.; Dickhout, J.G.; Ingram, A.J.; Austin, R.C. ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am. J. Physiol. Ren. Physiol. 2012, 303, F266–F278. [Google Scholar] [CrossRef]
- Escasany, E.; Izquierdo-Lahuerta, A.; Medina-Gomez, G. Underlying Mechanisms of Renal Lipotoxicity in Obesity. Nephron 2019, 143, 28–32. [Google Scholar] [CrossRef]
- Li, J.; Khodus, G.R.; Kruusmagi, M.; Kamali-Zare, P.; Liu, X.L.; Eklof, A.C.; Zelenin, S.; Brismar, H.; Aperia, A. Ouabain protects against adverse developmental programming of the kidney. Nat. Commun. 2010, 1, 42. [Google Scholar] [CrossRef]
- Hove-Madsen, L.; Llach, A.; Tort, L. Na(+)/Ca(2+)-exchange activity regulates contraction and SR Ca(2+) content in rainbow trout atrial myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1856–R1864. [Google Scholar] [CrossRef]
- Kumar, P.P.; Stotz, S.C.; Paramashivappa, R.; Beedle, A.M.; Zamponi, G.W.; Rao, A.S. Synthesis and evaluation of a new class of nifedipine analogs with T-type calcium channel blocking activity. Mol. Pharm. 2002, 61, 649–658. [Google Scholar] [CrossRef]
- Khan, K.M.; Patel, J.; Schaefer, T.J. Nifedipine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Shen, J.B.; Jiang, B.; Pappano, A.J. Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. J. Pharm. Exp. 2000, 294, 562–570. [Google Scholar]
- Murao, H.; Sakagami, N.; Iguchi, T.; Murakami, T.; Suketa, Y. Sodium fluoride increases intracellular calcium in rat renal epithelial cell line NRK-52E. Biol. Pharm. Bull. 2000, 23, 581–584. [Google Scholar] [CrossRef][Green Version]
- Bloch, K.M.; Evans, A.; Van Delft, J.; Lock, E.A. The effect of four non-genotoxic carcinogens and four non-carcinogens on NRK-52E cells using a transcriptomics approach. Toxicology 2011, 290, 110. [Google Scholar] [CrossRef]
- Altunkaynak, M.E.; Ozbek, E.; Altunkaynak, B.Z.; Can, I.; Unal, D.; Unal, B. The effects of high-fat diet on the renal structure and morphometric parametric of kidneys in rats. J. Anat. 2008, 212, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell. Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group/Parameter | Control | DR | DR + NF |
|---|---|---|---|
| Urinary protein (µg/dL) | 66.3 ± 8.8 a | 176.9 ± 13.2 b | 248.4 ± 10.8 c |
| Serum blood urea nitrogen/creatinine | 77.2 ± 5.4 a | 78.4 ± 10.0 a | 76. 3 ± 14.5 b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, C.-C.; Chen, C.-R.; Chen, C.-Y.; Lin, Y.-C.; Chen, K.-C.; Peng, R.Y. Nifedipine Upregulates ATF6-α, Caspases -12, -3, and -7 Implicating Lipotoxicity-Associated Renal ER Stress. Int. J. Mol. Sci. 2020, 21, 3147. https://doi.org/10.3390/ijms21093147
Peng C-C, Chen C-R, Chen C-Y, Lin Y-C, Chen K-C, Peng RY. Nifedipine Upregulates ATF6-α, Caspases -12, -3, and -7 Implicating Lipotoxicity-Associated Renal ER Stress. International Journal of Molecular Sciences. 2020; 21(9):3147. https://doi.org/10.3390/ijms21093147
Chicago/Turabian StylePeng, Chiung-Chi, Chang-Rong Chen, Chang-Yu Chen, Yen-Chung Lin, Kuan-Chou Chen, and Robert Y. Peng. 2020. "Nifedipine Upregulates ATF6-α, Caspases -12, -3, and -7 Implicating Lipotoxicity-Associated Renal ER Stress" International Journal of Molecular Sciences 21, no. 9: 3147. https://doi.org/10.3390/ijms21093147
APA StylePeng, C.-C., Chen, C.-R., Chen, C.-Y., Lin, Y.-C., Chen, K.-C., & Peng, R. Y. (2020). Nifedipine Upregulates ATF6-α, Caspases -12, -3, and -7 Implicating Lipotoxicity-Associated Renal ER Stress. International Journal of Molecular Sciences, 21(9), 3147. https://doi.org/10.3390/ijms21093147