Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
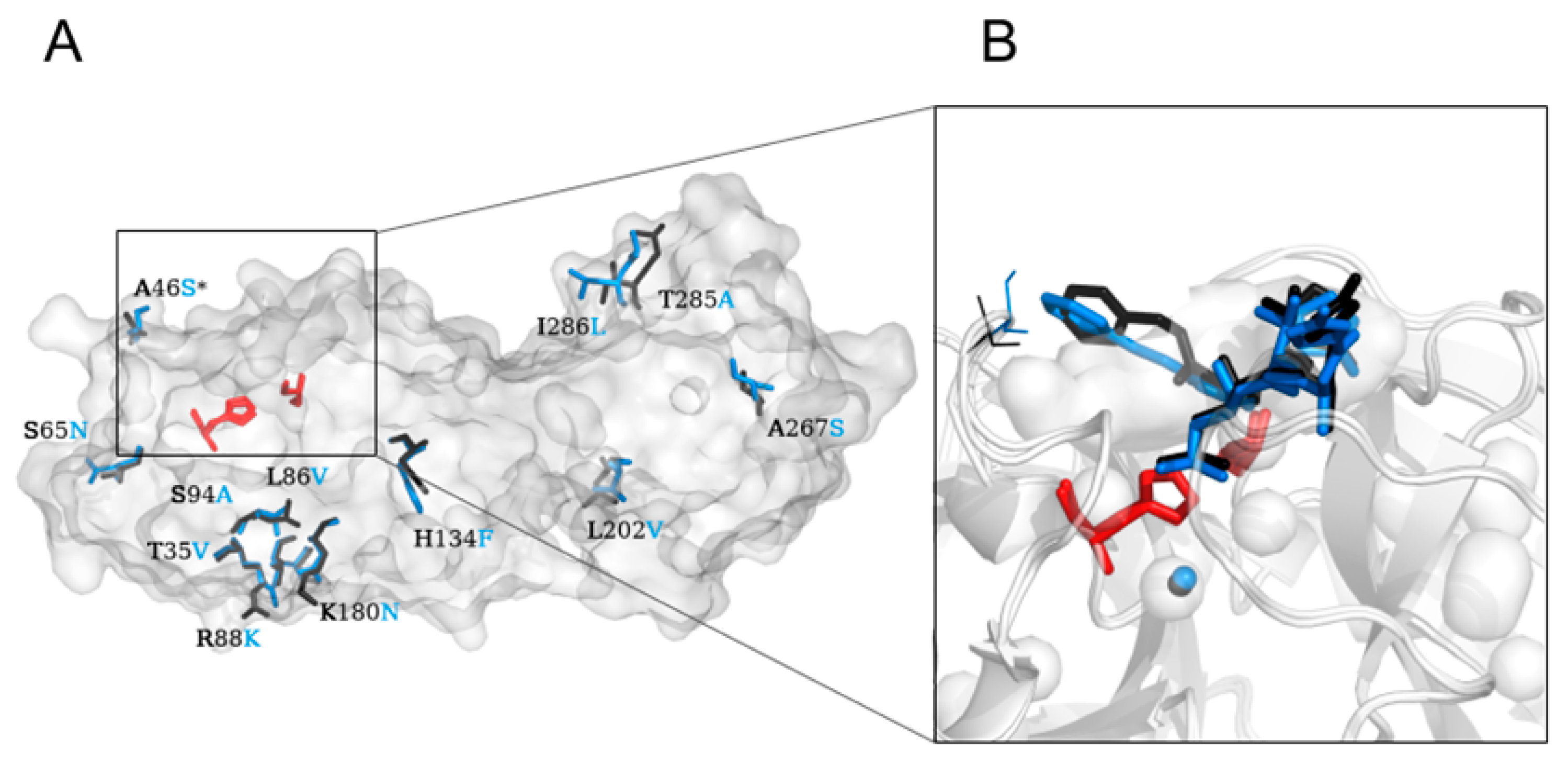
2.1. Crystal Structure Comparison, and Location of the Replaced Amino Acids Distal to the Active Site
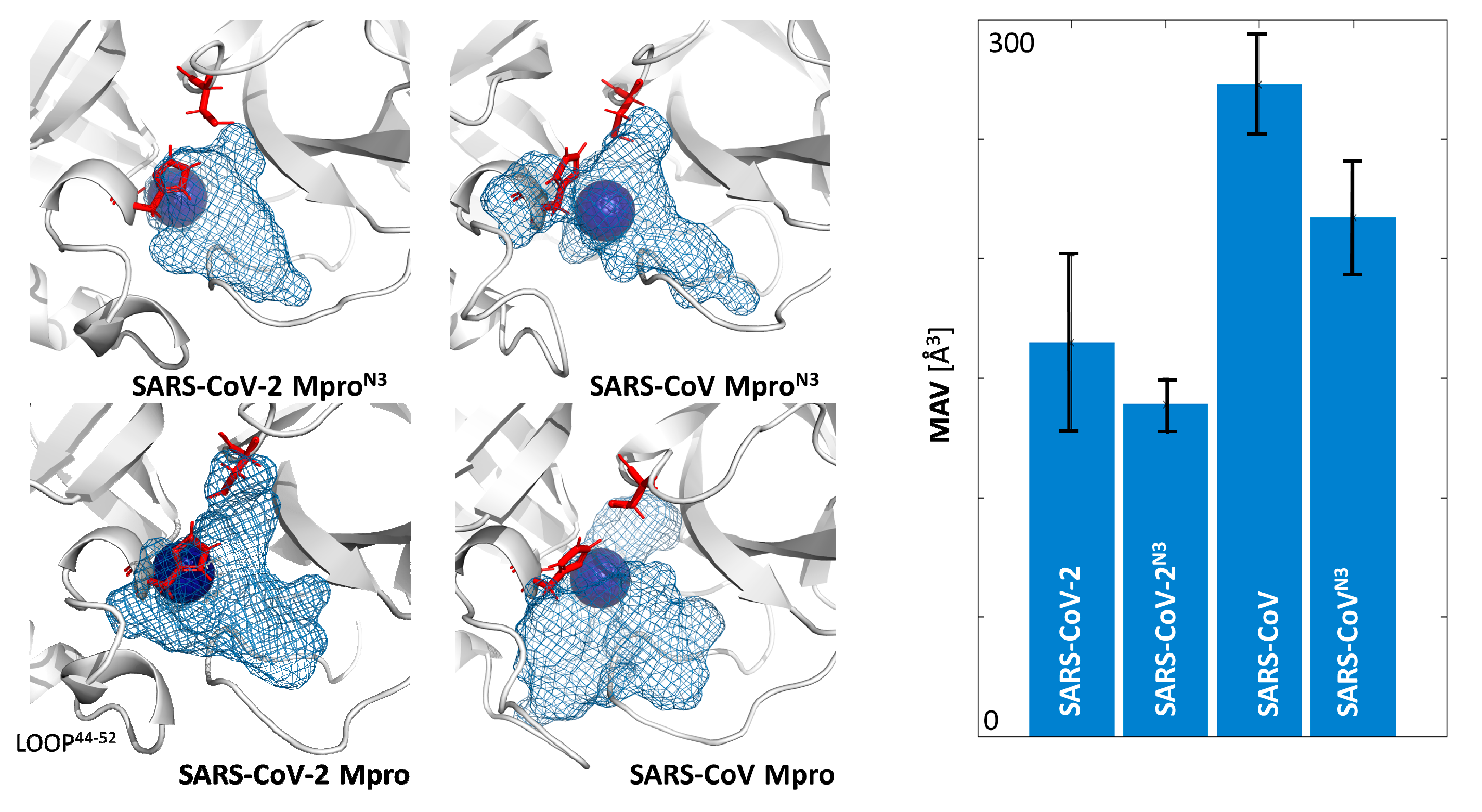
2.2. Plasticity of the Binding Cavities
2.3. Flexibility of the Active Site Entrance
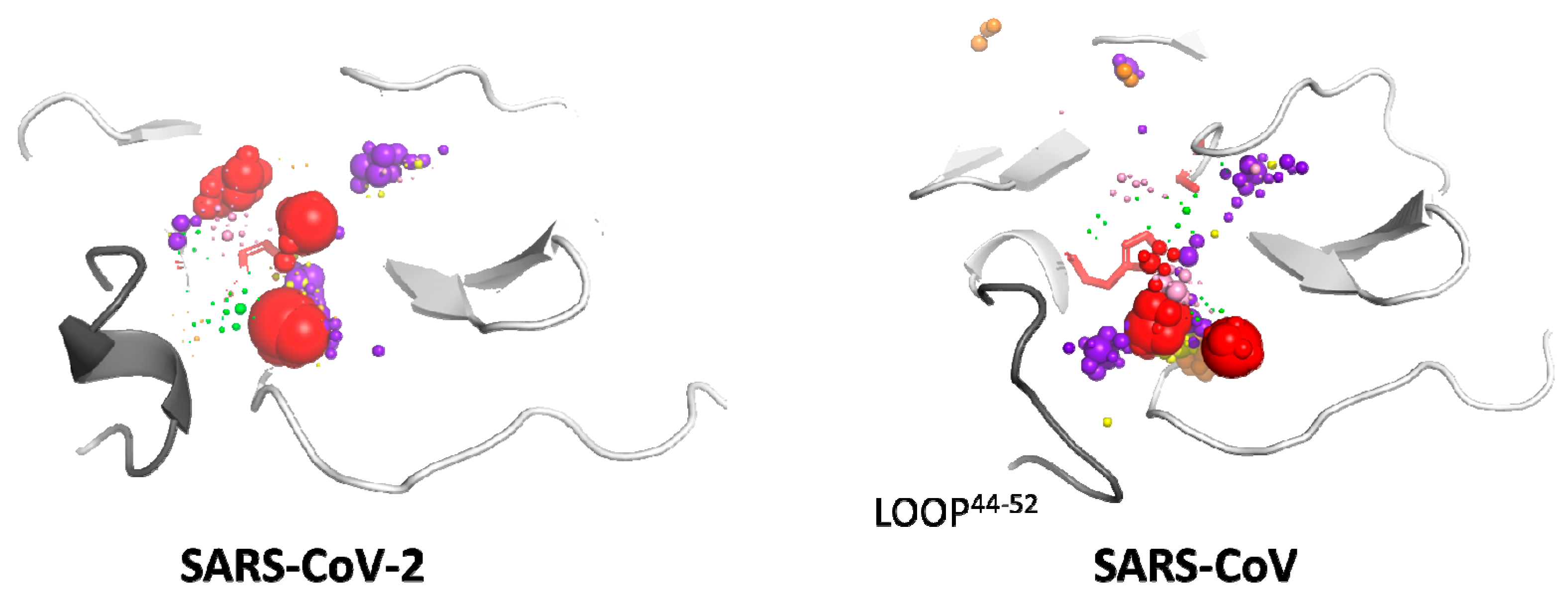
2.4. Cosolvent Hot-Spots Analysis
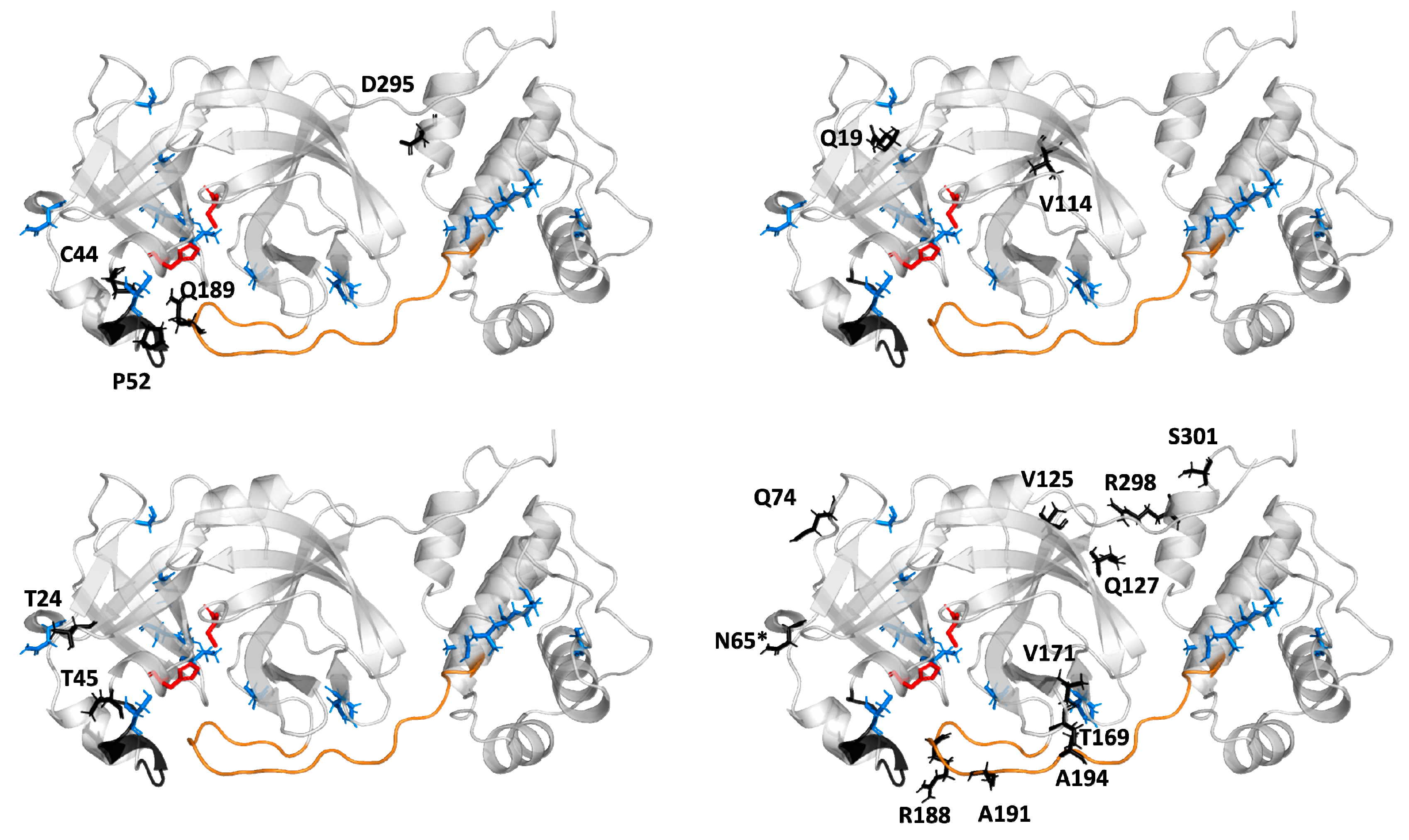
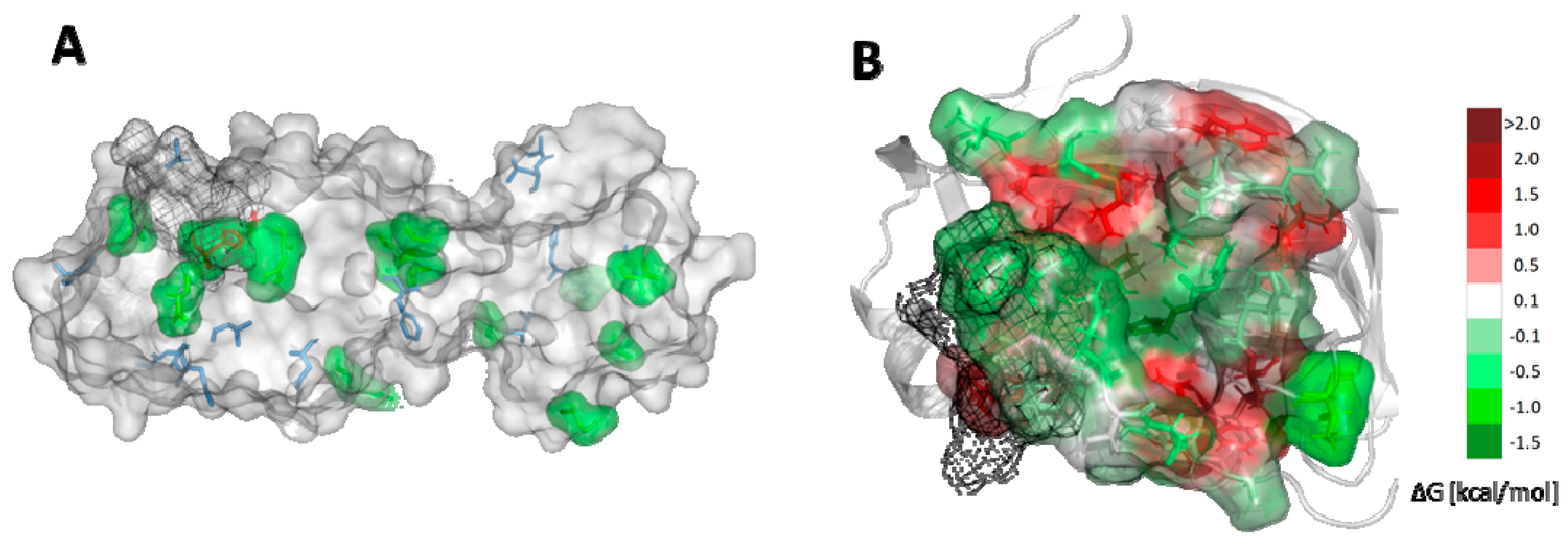
2.5. Potential Mutability of SARS-CoV-2
3. Discussion
4. Materials and Methods
4.1. Classical MD Simulations
4.2. Mixed-Solvent MD Simulations—Cosolvent Preparation
4.3. Mixed-Solvent MD Simulations—Initial Configuration
4.4. Water and Cosolvent Molecule Tracking
4.5. Hot-Spot Identification and Selection
4.6. Obtaining SARS-CoV-2 Mpro Gene Sequence
4.7. Energetic Effect of Amino Acid Substitutions
4.8. Comulator Calculations of Correlation Between Amino Acids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Mpro | Main protease |
| SARS | Severe acute respiratory syndrome |
| COVID-19 | Coronavirus disease 2019 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| CoVs | Coronaviruses |
| HCoV-NL63 | Human coronavirus NL63 |
| HCoV-229E | Human coronavirus 229E |
| HCoV-OC43 | Human coronavirus OC43 |
| HCoV-HKU1 | Human coronavirus HKU1 |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| ORFs | Open reading frames |
| 3CLpro | Chymotrypsin-like cysteine protease |
| S | Spike surface glycoprotein |
| E | Small envelope protein |
| M | Matrix protein |
| N | Nucleocapsid protein |
| N3(PRD_002214) | N-[(5 methylisoxazol-3-yl)carbonyl] alanyl-L-valyl-N~1-((1R,2Z)-4-(benzyloxy)-4-oxo-1-{[(3R)-2-oxopyrrolidin-3-yl]methyl}but-2-enyl)-L-leucinamide |
| cMD | Classical molecular dynamics simulations |
| MixMD | Mixed-solvent molecular dynamics simulations |
| PDB | Protein Data Bank |
| MAV | Maximal accessible volume |
| ACN | Acetonitrile |
| BNZ | Benzene |
| DMSO | Dimethylsulfoxide |
| MEO | Methanol |
| PHN | Phenol |
| URE | Urea |
| CMA | Correlated mutation analysis |
| FDA | Food and Drug Administration |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Song, Y.; Shi, M.; Cheng, Y.; Zhang, W.; Xia, X.-Q. Inferring the hosts of coronavirus using dual statistical models based on nucleotide composition. Sci. Rep. 2015, 5, 17155. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Zhang, L.; Shen, F.; Chen, F.; Lin, Z. Origin and evolution of the 2019 novel coronavirus. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Morse, J.S.; Lalonde, T.; Xu, S. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [Google Scholar]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Zhang, S.; Zou, P.; Chen, J.; Kang, X.; Li, Z.; Liang, C.; Jin, C.; Xia, B. Without Its N-Finger, the Main Protease of Severe Acute Respiratory Syndrome Coronavirus Can Form a Novel Dimer through Its C-Terminal Domain. J. Virol. 2008, 82, 4227–4234. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.-T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020. [Google Scholar] [CrossRef]
- Xu, Z.; Peng, C.; Shi, Y.; Zhu, Z.; Mu, K.; Wang, X.; Zhu, W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, X.; Wang, X.-J. Potential inhibitors for 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genomics 2020, 47, 119–121. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wang, N.; Li, H.; Shi, Y.; Guo, G.; Liu, K.; Hao, Z.; Zou, Q. Therapeutic Drugs Targeting 2019-nCoV Main Protease by High-Throughput Screening. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Gao, K.; Chen, J.; Wang, R.; Wei, G.-W. Potentially highly potent drugs for 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef]
- Talluri, S. Virtual High Throughput Screening Based Prediction of Potential Drugs for COVID-19. Preprints 2020. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sellner, M.; Neranjan, S.; Lill, M.A.; Smieško, M. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Inhibitors of SARS-CoV-2 Main Protease from a Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef]
- Adem, S.; Eyupoglu, V.; Sarfraz, I.; Rasul, A.; Ali, M. Identification of potent COVID-19 main protease (Mpro) inhibitors from natural polyphenols: An in silico strategy unveils a hope against CORONA. Preprints 2020. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Bacha, U.; Barrila, J.; Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Identification of Novel Inhibitors of the SARS Coronavirus Main Protease 3CL pro †. Biochemistry 2004, 43, 4906–4912. [Google Scholar] [CrossRef]
- Anand, K. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Mitusińska, K.; Raczyńska, A.; Bzówka, M.; Bagrowska, W.; Góra, A. Applications of water molecules for analysis of macromolecule properties. Comput. Struct. Biotechnol. J. 2020, 18, 355–365. [Google Scholar] [CrossRef]
- Magdziarz, T.; Mitusińska, K.; Bzówka, M.; Raczyńska, A.; Stańczak, A.; Banas, M.; Bagrowska, W.; Góra, A. AQUA-DUCT 1.0: Structural and functional analysis of macromolecules from an intramolecular voids perspective. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Li, C.; Qi, Y.; Teng, X.; Yang, Z.; Wei, P.; Zhang, C.; Tan, L.; Zhou, L.; Liu, Y.; Lai, L. Maturation Mechanism of Severe Acute Respiratory Syndrome (SARS) Coronavirus 3C-like Proteinase. J. Biol. Chem. 2010, 285, 28134–28140. [Google Scholar] [CrossRef]
- Panjkovich, A.; Daura, X. PARS: A web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics 2014, 30, 1314–1315. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, R.K.; Joosten, H.-J.; van Berkel, W.J.H.; Leferink, N.G.H.; Rooijen, E.; Ittmann, E.; van Zimmeren, F.; Jochens, H.; Bornscheuer, U.; Vriend, G.; et al. 3DM: Systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins Struct. Funct. Bioinform. 2010, 78, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Mitusińska, K.; Raedts, J.; Almourfi, F.; Joosten, H.-J.; Hendriks, S.; Sedelnikova, S.E.; Kengen, S.W.M.; Hagen, W.R.; Góra, A.; et al. Distant Non-Obvious Mutations Influence the Activity of a Hyperthermophilic Pyrococcus furiosus Phosphoglucose Isomerase. Biomolecules 2019, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins Struct. Funct. Bioinform. 2019, 87, 520–527. [Google Scholar] [CrossRef]
- Spyrakis, F.; Ahmed, M.H.; Bayder, A.S.; Cozzini, P.; Mozzarelli, A.; Kellogg, G.E. The Roles of Water in the Protein Matrix: A Largely Untapped Resource for Drug Discovery. J. Med. Chem. 2017, 60, 6781–6827. [Google Scholar] [CrossRef]
- De Beer, S.B.A.; Vermeulen, N.P.E.; Oostenbrink, C. The Role of Water Molecules in Computational Drug Design. Curr. Top. Med. Chem. 2010, 10, 55–66. [Google Scholar] [CrossRef]
- Mitusińska, K.; Magdziarz, T.; Bzówka, M.; Stańczak, A.; Gora, A. Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking. Biomolecules 2018, 8, 143. [Google Scholar] [CrossRef]
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1102–1111. [Google Scholar] [CrossRef]
- Tsai, M.-Y.; Chang, W.-H.; Liang, J.-Y.; Lin, L.-L.; Chang, G.-G.; Chang, H.-P. Essential covalent linkage between the chymotrypsin-like domain and the extra domain of the SARS-CoV main protease. J. Biochem. 2010, 148, 349–358. [Google Scholar] [CrossRef]
- Anand, K. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; Shi, J.; Mu, Y.; Song, J. Dynamically-Driven Enhancement of the Catalytic Machinery of the SARS 3C-Like Protease by the S284-T285-I286/A Mutations on the Extra Domain. PLoS ONE 2014, 9, e101941. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Jeyachandran, S.; Hu, N.-J.; Liu, C.-L.; Lin, S.-Y.; Wang, Y.-S.; Chang, Y.-M.; Hou, M.-H. Structure-based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein. Mol. Biosyst. 2016, 12, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Dayer, M.R.; Taleb-Gassabi, S.; Dayer, M.S. Lopinavir; A Potent Drug against Coronavirus Infection: Insight from Molecular Docking Study. Arch. Clin. Infect. Dis. 2017, 12. [Google Scholar] [CrossRef]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Luchko, T.; Gusarov, S.; Roe, D.R.; Simmerling, C.; Case, D.A.; Tuszynski, J.; Kovalenko, A. Three-Dimensional Molecular Theory of Solvation Coupled with Molecular Dynamics in Amber. J. Chem. Theory Comput. 2010, 6, 607–624. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution-Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham III, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Heo, L.; Feig, M. What makes it difficult to refine protein models further via molecular dynamics simulations? Proteins Struct. Funct. Bioinform. 2018, 86, 177–188. [Google Scholar] [CrossRef]
- Mitusińska, K.; Skalski, T.; Góra, A. Simple selection procedure to distinguish between static and flexible loops. Int. J. Mol. Sci. 2020, 21, 2293. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Nikitin, A.M.; Lyubartsev, A.P. New six-site acetonitrile model for simulations of liquid acetonitrile and its aqueous mixtures. J. Comput. Chem. 2007, 28, 2020–2026. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger; Schrödinger LLC.: New York, NY, USA.
- Gertz, E.M.; Yu, Y.-K.; Agarwala, R.; Schäffer, A.A.; Altschul, S.F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 2006, 4, 41. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. Methods Mol. Biol. 2014, 1079, 105–106. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099. https://doi.org/10.3390/ijms21093099
Bzówka M, Mitusińska K, Raczyńska A, Samol A, Tuszyński JA, Góra A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. International Journal of Molecular Sciences. 2020; 21(9):3099. https://doi.org/10.3390/ijms21093099
Chicago/Turabian StyleBzówka, Maria, Karolina Mitusińska, Agata Raczyńska, Aleksandra Samol, Jack A. Tuszyński, and Artur Góra. 2020. "Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design" International Journal of Molecular Sciences 21, no. 9: 3099. https://doi.org/10.3390/ijms21093099
APA StyleBzówka, M., Mitusińska, K., Raczyńska, A., Samol, A., Tuszyński, J. A., & Góra, A. (2020). Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. International Journal of Molecular Sciences, 21(9), 3099. https://doi.org/10.3390/ijms21093099