Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies

 , , ,
, , , 
{kind=link}
{kind=link}
Abstract
1. Introduction
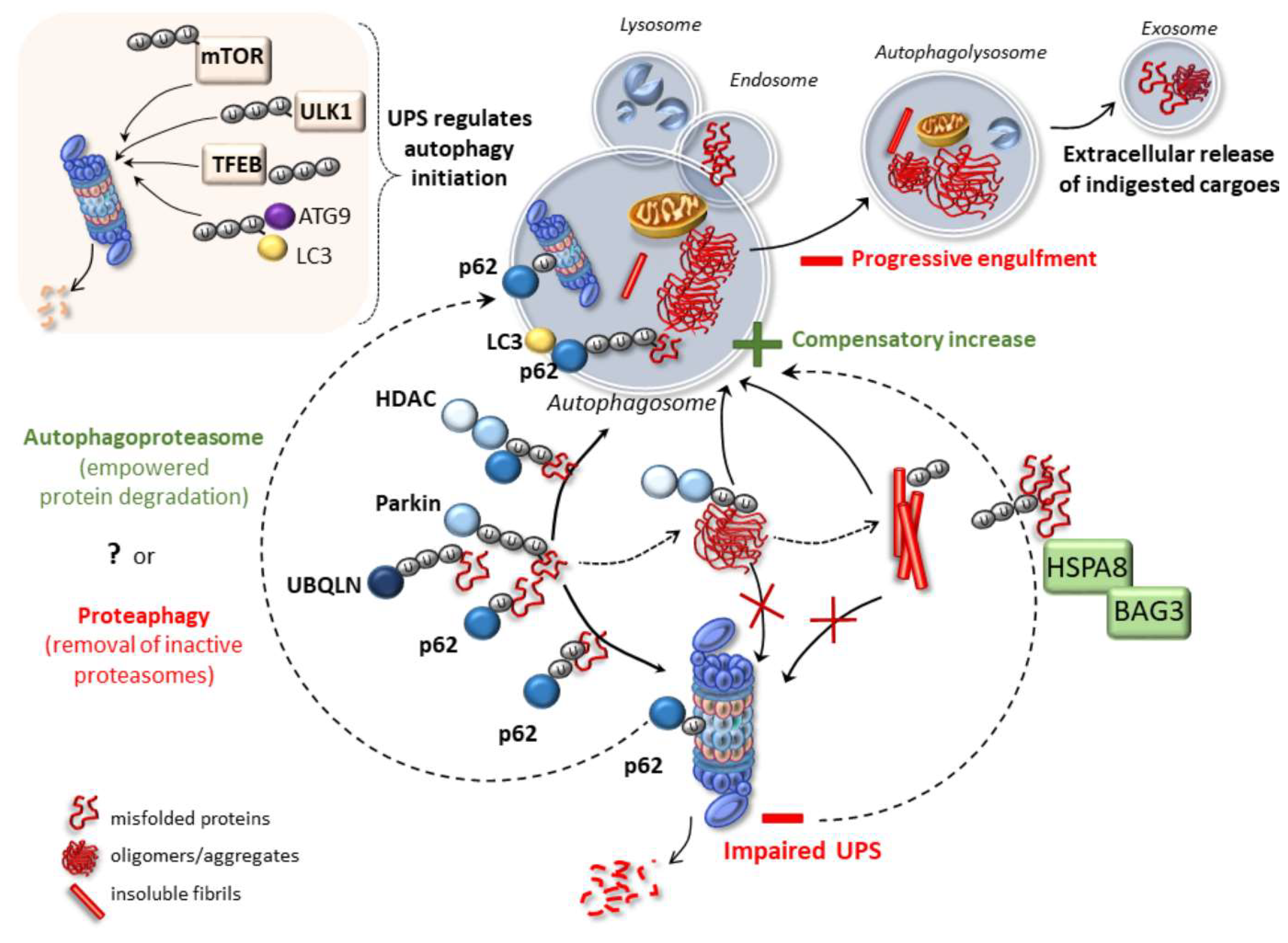
2. Emerging Mechanisms Underlying Autophagy and Proteasome Cross-Talk in Health and Disease
3. Autophagy and Proteasome Interplay in Prion-Like Protein Clearance
3.1. Alpha-Synuclein
3.2. Amyloid Beta and tau
3.3. Huntingtin
3.4. TDP-43
3.5. FUS
3.6. SOD1
4. Cell-Clearing Systems and Prion-Like Protein Propagation
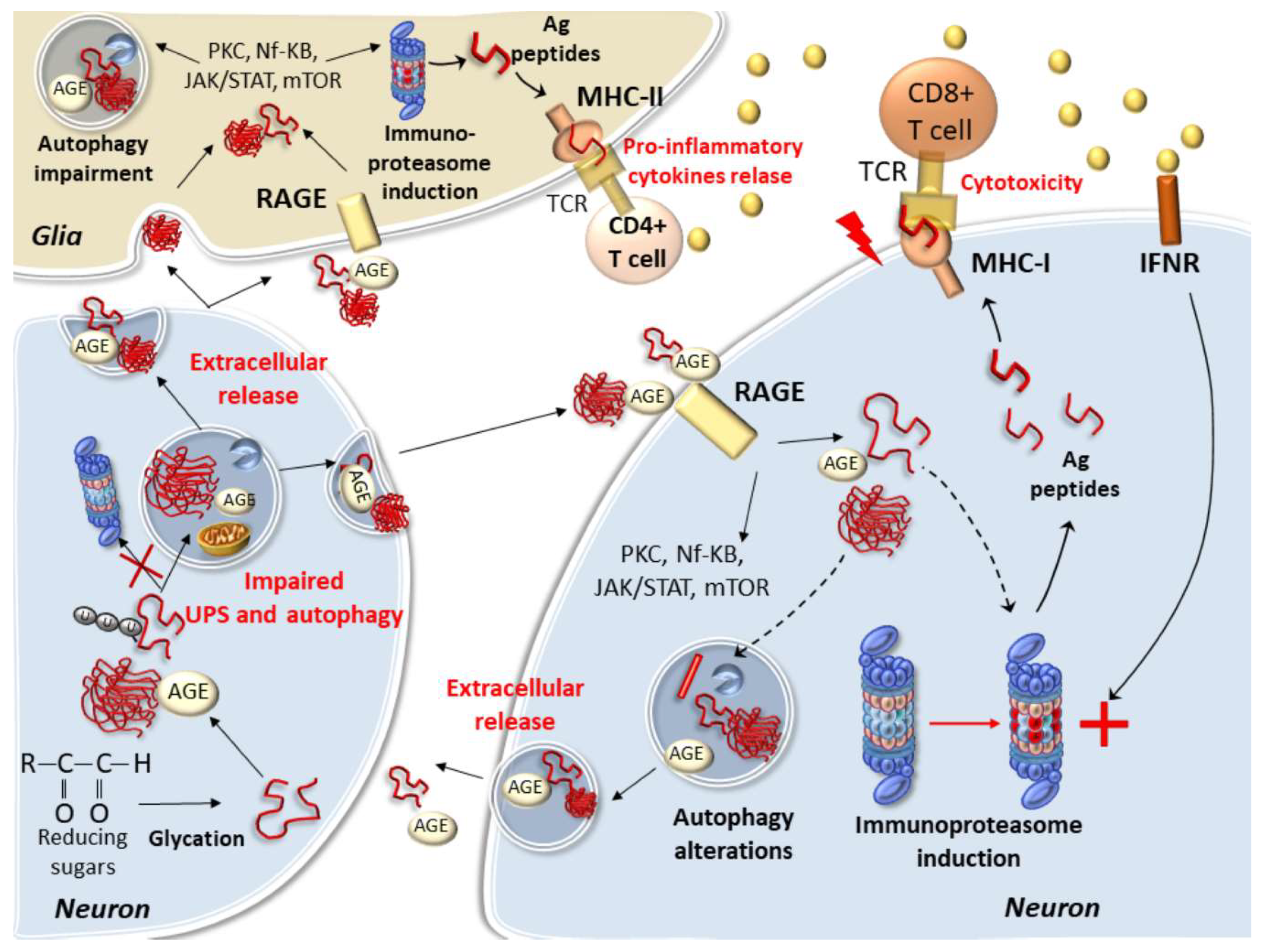
5. Protein Glycation and Cell-Clearing Systems Alterations Bridging Cell-to-Cell Propagation and Neuro-Inflammation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| HD | Huntington Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| FTD | Frontotemporal Dementia |
| MSA | Multiple System Atrophy |
| α-syn | Alpha-Synuclein |
| Aβ | Amyloid-Beta |
| AβPP | Amyloid-Beta Precursor Protein |
| TDP-43 | Tar-DNA-Binding Protein of 43 KDa |
| SOD | Cu/Zn Superoxide Dismutase |
| FUS | Fused In Sarcoma |
| UPS | Ubiquitin Proteasome System |
| UBQLNs | Ubiquilin Proteins |
| HDAC6 | Histone Deacetylase 6 |
| UBA | Ubiquitin-Associated |
| UBL | Ubiquitin-Ligated |
| UBA6 | Ubiquitin-Activating Enzyme (E1) |
| BIRC6 | Hybrid Ubiquitin-Conjugating Enzyme/Ubiquitin-Ligase (E2/E3) |
| GABARAP | GABA Type A Receptor-Associated Protein |
| METH | Methamphetamine |
| mTOR | Mammalian/Mechanistic Target Of Rapamycin |
| TFEB | Transcription Factor EB |
| FBXO7 | F-box only protein 7 |
| LAMP1 | Lysosomal-Associated Membrane Protein 1 |
| LC3 | Microtubule-Associated Protein 1a/1b-Light Chain 3 |
| HERC1 | HECT and RLD Domain Containing E3 Ubiquitin Protein Ligase Family Member 1 |
| HSPA8 | Heat Shock Protein Family A (Hsp70) Member 8 |
| HSPA1A | Heat Shock Protein Family A (Hsp70) Member 1A |
| BAG1/3 | Bcl2-Associated Athanogene 1/3 |
| DLB | Dementia with Lewy Bodies |
| PSP | Progressive Supranuclear Palsy |
| MPTP | 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine |
| 6-OHDA | 6-Hydroxydopamine |
| 3-MA | 3-Methyladenine |
| USP9X | Ubiquitin Specific Peptidase 9 X-Linked |
| APP | Amyloid Precursor Protein |
| GSK3β | Glycogen Synthase Kinase3 Beta |
| BACE2 | Beta-Secretase 2 |
| POLDIP2 | DNA Polymerase Delta Interacting Protein 2 |
| NUB1 | Negative Regulator of Ubiquitin-Like Protein 1 |
| USP14 | Ubiquitin Specific Peptidase 14 |
| HSC70 | Heat Shock Cognate 71 Kda Protein (Hsc70), also known as HSPA8 |
| EVs | Extracellular Vesicles |
| CSF | Cerebrospinal Fluid |
| CNS | Central Nervous System |
| AGEs | Advanced Glycation End Products |
| RAGEs | Receptors for Advanced Glycation End Products |
| PKC | Protein Kinase C |
| NF-kB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| JAK2/STAT1 | Janus Kinase 2/Signal Transducer and Activator of Transcription 1 |
| Ag | Antigen |
| MHC-I | Major Histocompatibility Complex Type I |
| MHC-II | Major Histocompatibility Complex Type II |
| CD8+ T-cells | CD8 positive Cytotoxic T Lymphocytes |
| CD4+ Th cells | CD4 positive T Helper Lymphocytes |
| TLR4 | Toll-Like Receptor 4 |
| ULK1 | Unc-51-Like Autophagy-Activating Kinase 1 |
References
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Hanni, K.B.; Marksberry, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.E.; Orr, A.; Tydlacka, S.; Li, S.H.; Li, X.J. Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J. Cell Biol. 2008, 180, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.S. Proteasome degradation of brain cytosolic tau in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2011, 4, 385–402. [Google Scholar]
- Graham, S.H.; Liu, H. Life and death in the trash heap: The ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral Ischemia. Ageing Res. Rev. 2017, 34, 30–38. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Kroemer, G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009, 23, 2253–2259. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Gambardella, S.; Frati, A.; Fornai, F. Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia. Int. J. Mol. Sci. 2018, 19, 2756. [Google Scholar] [CrossRef]
- Ryskalin, L.; Busceti, C.L.; Limanaqi, F.; Biagioni, F.; Gambardella, S.; Fornai, F. A Focus on the Beneficial Effects of Alpha Synuclein and a Re-Appraisal of Synucleinopathies. Curr. Protein Pept. Sci. 2018, 19, 598–611. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Ryskalin, L.; Ruffoli, R.; Biagioni, F.; Limanaqi, F.; Ferrucci, M.; Busceti, C.L.; Bonuccelli, U.; Fornai, F. The Neuroanatomy of the Reticular Nucleus Locus Coeruleus in Alzheimer’s Disease. Front. Neuroanat. 2017, 11, 80. [Google Scholar] [CrossRef]
- Ferrucci, M.; Fulceri, F.; Toti, L.; Soldani, P.; Siciliano, G.; Paparelli, A.; Fornai, F. Protein clearing pathways in ALS. Arch. Ital. Biol. 2011, 149, 121–149. [Google Scholar]
- Kovacs, G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef]
- Jellinger, K.A. Interaction between pathogenic proteins in neurodegenerative disorders. J. Cell. Mol. Med. 2012, 16, 1166–1183. [Google Scholar] [CrossRef] [PubMed]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis [published correction appears in Front. Mol. Neurosci. 2020, 12, 311]. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Gelpi, E.; Charif, S.; Belbin, O.; Blesa, R.; Martí, M.J.; Clarimón, J.; Lleó, A. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2013, 72, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Charles, V.; Mezey, E.; Reddy, P.H.; Dehejia, A.; Young, T.A.; Polymeropoulos, M.H.; Brownstein, M.J.; Tagle, D.A. α-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci. Lett. 2000, 289, 29–32. [Google Scholar] [CrossRef]
- Takei, Y.; Oguchi, K.; Koshihara, H.; Hineno, A.; Nakamura, A.; Ohara, S. α-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Hum. Pathol. 2013, 44, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef]
- Pasquali, L.; Lenzi, P.; Biagioni, F.; Siciliano, G.; Fornai, F. Cell to cell spreading of misfolded proteins as a therapeutic target in motor neuron disease. Curr. Med. Chem. 2014, 21, 3508–3534. [Google Scholar] [CrossRef]
- Nonaka, T. The Propagation Hypothesis of Prion-like Protein Agregates in Neurodegenerative Diseases. Brain Nerve 2019, 71, 1209–1214. [Google Scholar]
- Hasegawa, M.; Nonaka, T.; Masuda-Suzukake, M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol. Ther. 2017, 172, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Jucker, M.; Walker, L.C. Aβ seeds and prions: How close the fit? Prion 2017, 11, 215–225. [Google Scholar] [CrossRef] [PubMed]
- McAllister, B.B.; Lacoursiere, S.G.; Sutherland, R.J.; Mohajerani, M.H. Intracerebral seeding of amyloid-β and tau pathology in mice: Factors underlying prion-like spreading and comparisons with α-synuclein. Neurosci. Biobehav. Rev. 2020, 112, 1–27. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell. Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4–E6; discussion E6–7. [Google Scholar]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2016, 139, 481–494. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Udan, M.; Baloh, R.H. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 2011, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of beta-amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Lécolle, K.; Caillierez, R.; Bégard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. Handb. Clin. Neurol. 2018, 153, 303–319. [Google Scholar]
- Masnata, M.; Sciacca, G.; Maxan, A.; Bousset, L.; Denis, H.L.; Lauruol, F.; David, L.; Saint-Pierre, M.; Kordower, J.H.; Melki, R.; et al. Demonstration of prion-like properties of mutant huntingtin fibrils in both in vitro and in vivo paradigms. Acta Neuropathol. 2019, 137, 981–1001. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell. Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.C.; Bishop, J.F.; Leng, Y.; Chock, P.B.; Chase, T.N.; Mouradian, M.M. Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 1999, 274, 33855–33858. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Polzella, M.; Frati, A.; Fornai, F. Phytochemicals Bridging Autophagy Induction and Alpha-Synuclein Degradation in Parkinsonism. Int. J. Mol. Sci. 2019, 20, 3274. [Google Scholar] [CrossRef]
- Lee, S.; Jeon, Y.M.; Cha, S.J.; Kim, S.; Kwon, Y.; Jo, M.; Jang, Y.N.; Lee, S.; Kim, J.; Kim, S.R.; et al. PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 2019, 1–17. [Google Scholar] [CrossRef]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef]
- Zhao, J.; Goldberg, A.L. Coordinate regulation of autophagy and the ubiquitin proteasome system by MTOR. Autophagy 2016, 12, 1967–1970. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62-and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 2016, 7, 83833–83834. [Google Scholar] [CrossRef]
- Kageyama, S.; Sou, Y.S.; Uemura, T.; Kametaka, S.; Saito, T.; Ishimura, R.; Kouno, T.; Bedford, L.; Mayer, R.J.; Lee, M.S.; et al. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J. Biol. Chem. 2014, 289, 24944–24955. [Google Scholar] [CrossRef]
- Yang, F.; Yang, Y.P.; Mao, C.J.; Liu, L.; Zheng, H.F.; Hu, L.F.; Liu, C.F. Crosstalk between the proteasome system and autophagy in the clearance of α-synuclein. Acta Pharmacol. Sin. 2013, 34, 674–680. [Google Scholar] [CrossRef]
- Agholme, L.; Hallbeck, M.; Benedikz, E.; Marcusson, J.; Kågedal, K. Amyloid-β secretion, generation, and lysosomal sequestration in response to proteasome inhibition: Involvement of autophagy. J. Alzheimers Dis. 2012, 31, 343–358. [Google Scholar] [CrossRef]
- Ugun-Klusek, A.; Tatham, M.H.; Elkharaz, J.; Constantin-Teodosiu, D.; Lawler, K.; Mohamed, H.; Paine, S.M.; Anderson, G.; John Mayer, R.; Lowe, J.; et al. Continued 26S proteasome dysfunction in mouse brain cortical neurons impairs autophagy and the Keap1-Nrf2 oxidative defence pathway. Cell Death Dis. 2017, 8, e2531. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef] [PubMed]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-to-autophagy switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [PubMed]
- Cecarini, V.; Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Rossi, G.; Buizza, L.; Uberti, D.; Angeletti, M.; Eleuteri, A.M. Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Soldani, P.; Frati, A.; Fornai, F. Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity. Int. J. Mol. Sci. 2019, 20, 2197. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef]
- Qin, Z.H.; Wang, Y.M.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.B.; Menendez-Benito, V.; Dantuma, N.P. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell. 2007, 26, 175–188. [Google Scholar] [CrossRef]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Lan, D.; Wang, W.; Zhuang, J.; Zhao, Z. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant α-synuclein. Mol. Med. Rep. 2015, 11, 1655–1660. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Harman, C.A.; Monteiro, M.J. The specificity of ubiquitin binding to ubiquilin-1 is regulated by sequences besides its UBA domain. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Şentürk, M.; Lin, G.; Zuo, Z.; Mao, D.; Watson, E.; Mikos, A.G.; Bellen, H.J. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol. 2019, 21, 384–396. [Google Scholar] [CrossRef]
- Kim, S.H.; Stiles, S.G.; Feichtmeier, J.M.; Ramesh, N.; Zhan, L.; Scalf, M.A.; Smith, L.M.; Pandey, U.B.; Tibbetts, R.S. Mutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS. Hum. Mol. Genet. 2018, 27, 322–337. [Google Scholar] [CrossRef]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef]
- Cakir, Z.; Funk, K.; Lauterwasser, J.; Todt, F.; Zerbes, R.M.; Oelgeklaus, A.; Tanaka, A.; van der Laan, M.; Edlich, F. Parkin promotes proteasomal degradation of misregulated BAX. J. Cell Sci. 2017, 130, 2903–2913. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012, 287, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar]
- Otero, M.G.; Alloatti, M.; Cromberg, L.E.; Almenar-Queralt, A.; Encalada, S.E.; Pozo Devoto, V.M.; Bruno, L.; Goldstein, L.S.; Falzone, T.L. Fast axonal transport of the proteasome complex depends on membrane interaction and molecular motor function. J. Cell. Sci. 2014, 127, 1537–1549. [Google Scholar] [CrossRef]
- Gao, G.; He, J.; Luo, Y.; Sun, Y.; Zhou, Y.; Zhang, J.; Xing, Y.; Dai, J. Axonopathy Likely Initiates Neuropathological Processes Via a Mechanism of Axonal Leakage in Alzheimer’s Mouse Models. Curr. Mol. Med. 2019, 19, 183–195. [Google Scholar] [CrossRef]
- Ojelade, S.A.; Lee, T.V.; Giagtzoglou, N.; Yu, L.; Ugur, B.; Li, Y.; Duraine, L.; Zuo, Z.; Petyuk, V.; De Jager, P.L.; et al. cindr, the Drosophila Homolog of the CD2AP Alzheimer’s Disease Risk Gene, Is Required for Synaptic Transmission and Proteostasis. Cell Rep. 2019, 28, 1799–1813. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Ryskalin, L.; Biagioni, F.; Gambardella, S.; Busceti, C.L.; Falleni, A.; Lazzeri, G.; Fornai, F. Methamphetamine increases Prion Protein and induces dopamine-dependent expression of protease resistant PrPsc. Arch. Ital. Biol. 2017, 155, 81–97. [Google Scholar] [PubMed]
- Biagioni, F.; Ferese, R.; Limanaqi, F.; Madonna, M.; Lenzi, P.; Gambardella, S.; Fornai, F. Methamphetamine persistently increases alpha-synuclein and suppresses gene promoter methylation within striatal neurons. Brain Res. 2019, 1719, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gaglione, A.; Busceti, C.L.; Fornai, F. A Sentinel in the Crosstalk Between the Nervous and Immune System: The (Immuno)-Proteasome. Front. Immunol. 2019, 10, 628. [Google Scholar] [CrossRef]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol. Cell. 2016, 61, 625–639. [Google Scholar] [CrossRef]
- Broekaart, D.W.M.; van Scheppingen, J.; Geijtenbeek, K.W.; Zuidberg, M.R.J.; Anink, J.J.; Baayen, J.C.; Mühlebner, A.; Aronica, E.; Gorter, J.A.; van Vliet, E.A. Increased expression of (immuno)proteasome subunits during epileptogenesis is attenuated by inhibition of the mammalian target of rapamycin pathway. Epilepsia 2017, 58, 1462–1472. [Google Scholar] [CrossRef]
- Marshall, R.S.; Li, F.; Gemperline, D.C.; Book, A.J.; Vierstra, R.D. Autophagic Degradation of the 26S Proteasome Is Mediated by the Dual ATG8/Ubiquitin Receptor RPN10 in Arabidopsis. Mol. Cell. 2015, 58, 1053–1066. [Google Scholar] [CrossRef]
- Nazio, F.; Carinci, M.; Valacca, C.; Bielli, P.; Strappazzon, F.; Antonioli, M.; Ciccosanti, F.; Rodolfo, C.; Campello, S.; Fimia, G.M.; et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 2016, 215, 841–856. [Google Scholar] [CrossRef]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol. Cell. 2016, 61, 84–97. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Li, X.; Qiu, K.; Jiao, F.; Liu, Y.; Kong, Q.; Liu, Y.; Wu, Y. Proteasome Inhibition Activates Autophagy-Lysosome Pathway Associated with TFEB Dephosphorylation and Nuclear Translocation. Front. Cell. Dev. Biol. 2019, 7, 170. [Google Scholar] [CrossRef]
- Follo, C.; Vidoni, C.; Morani, F.; Ferraresi, A.; Seca, C.; Isidoro, C. Amino acid response by Halofuginone in Cancer cells triggers autophagy through proteasome degradation of mTOR. Cell Commun. Signal. 2019, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Rios, L.; Yan, Z.; Jasper, A.M.; Luera, D.; Luo, S.; Rao, H. Autophagy regulator Atg9 is degraded by the proteasome. Biochem. Biophys. Res. Commun. 2020, 522, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Papinski, D.; Kraft, C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J. Mol. Biol. 2016, 428, 1725–1741. [Google Scholar] [CrossRef]
- Jia, R.; Bonifacino, J.S. Negative regulation of autophagy by UBA6-BIRC6-mediated ubiquitination of LC3. Elife 2019, 8, e50034. [Google Scholar] [CrossRef]
- Jia, R.; Bonifacino, J.S. Regulation of LC3B levels by ubiquitination and proteasomal degradation. Autophagy 2020, 16, 382–384. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Ruffoli, R.; Soldani, P.; Ruggieri, S.; Alessandri, M.G.; et al. Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J. Neurosci. 2003, 23, 8955–8966. [Google Scholar] [CrossRef]
- Fornai, F.; Lazzeri, G.; Bandettini Di Poggio, A.; Soldani, P.; De Blasi, A.; Nicoletti, F.; Ruggieri, S.; Paparelli, A. Convergent roles of alpha-synuclein, DA metabolism, and the ubiquitin-proteasome system in nigrostriatal toxicity. Ann. N. Y. Acad. Sci. 2006, 1074, 84–89. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef]
- Sato, S.; Hattori, N. Dopaminergic Neuron-Specific Autophagy-Deficient Mice. Methods Mol. Biol. 2018, 1759, 173–175. [Google Scholar] [PubMed]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Hadjebi, O.; Amair-Pinedo, F.; Tsurumi, T.; Langa, F.; Serikawa, T.; Sotelo, C.; Guénet, J.L.; Rosa, J.L. Progressive Purkinje cell degeneration in tambaleante mutant mice is a consequence of a missense mutation in HERC1 E3 ubiquitin ligase. PLoS Genet. 2009, 5, e1000784. [Google Scholar] [CrossRef]
- Ruiz, R.; Pérez-Villegas, E.M.; Bachiller, S.; Rosa, J.L.; Armengol, J.A. HERC 1 Ubiquitin Ligase Mutation Affects Neocortical, CA3 Hippocampal and Spinal Cord Projection Neurons: An Ultrastructural Study. Front. Neuroanat. 2016, 10, 42. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Kasten, M.; Klein, C. The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. French Parkinson’s Disease Genetics Study Group. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, S.; Ferese, R.; Biagioni, F.; Busceti, C.L.; Campopiano, R.; Griguoli, A.M.P.; Limanaqi, F.; Novelli, G.; Storto, M.; Fornai, F. The Monoamine Brainstem Reticular Formation as a Paradigm for Re-Defining Various Phenotypes of Parkinson’s Disease Owing Genetic and Anatomical Specificity. Front. Cell. Neurosci. 2017, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol. 2012, 124, 643–664. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Dementia with Lewy bodies and Parkinson’s disease-dementia: Current concepts and controversies. J. Neural Transm. 2018, 125, 615–650. [Google Scholar] [CrossRef]
- Tong, J.; Wong, H.; Guttman, M.; Ang, L.C.; Forno, L.S.; Shimadzu, M.; Rajput, A.H.; Muenter, M.D.; Kish, S.J.; Hornykiewicz, O.; et al. Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: A comparative investigation. Brain 2010, 133, 172–188. [Google Scholar] [CrossRef]
- Zhao, H.Q.; Li, F.F.; Wang, Z.; Wang, X.M.; Feng, T. A comparative study of the amount of α-synuclein in ischemic stroke and Parkinson’s disease. Neurol. Sci. 2016, 37, 749–754. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; de Blasi, A.; Battaglia, G.; Nicoletti, F.; Ruggieri, S.; et al. Similarities between methamphetamine toxicity and proteasome inhibition. Ann. N.Y. Acad. Sci. 2004, 1025, 162–170. [Google Scholar] [CrossRef]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Chen, B.; Zhang, J.P.; Ma, Y.Y. Up-regulation of autophagy-related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson’s disease. J. Biol Chem. 2017, 292, 18062–18074. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Höglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the striatal 6-OHDA model of Parkinson’s disease in wild type and alpha-synuclein-deleted mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Alves da Costa, C.; Dunys, J.; Brau, F.; Wilk, S.; Cappai, R.; Checler, F. 6-Hydroxydopamine but not 1-methyl-4-phenylpyridinium abolishes alpha-synuclein anti-apoptotic phenotype by inhibiting its proteasomal degradation and by promoting its aggregation. J. Biol. Chem. 2006, 281, 9824–9831. [Google Scholar] [CrossRef]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Rideout, H.J.; Larsen, K.E.; Sulzer, D.; Stefanis, L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J. Neurochem. 2001, 78, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Bandettini di Poggio, A.; Battaglia, G.; Pellegrini, A.; Murri, L.; Ruggieri, S.; Paparelli, A.; Fornai, F. A short overview on the role of alpha-synuclein and proteasome in experimental models of Parkinson’s disease. J. Neural Transm. Suppl. 2006, 70, 105–109. [Google Scholar]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001, 509, 22–26. [Google Scholar] [CrossRef]
- Batelli, S.; Peverelli, E.; Rodilossi, S.; Forloni, G.; Albani, D. Macroautophagy and the proteasome are differently involved in the degradation of alpha-synuclein wild type and mutated A30P in an in vitro inducible model (PC12/TetOn). Neuroscience 2011, 195, 128–137. [Google Scholar] [CrossRef][Green Version]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef]
- Yan, J.Q.; Yuan, Y.H.; Chu, S.F.; Li, G.H.; Chen, N.H. E46K Mutant α-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway. Molecules 2018, 23, 2839. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S. α-Synuclein fate: Proteasome or autophagy? Autophagy 2012, 8, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Benjamin, S.; Wapinski, O.L.; Smith, D.M.; Goldberg, A.L.; Steller, H. A conserved F box regulatory complex controls proteasome activity in Drosophila. Cell 2011, 145, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Severijnen, L.A.; van der Weiden, M.; Zheng, P.P.; Oostra, B.A.; Hukema, R.K.; Willemsen, R.; Kros, J.M.; Bonifati, V. FBXO7 immunoreactivity in α-synuclein-containing inclusions in Parkinson disease and multiple system atrophy. J. Neuropathol. Exp. Neurol. 2013, 72, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.; Tang, A.M.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef]
- Teixeira, F.R.; Randle, S.J.; Patel, S.P.; Mevissen, T.E.; Zenkeviciute, G.; Koide, T.; Komander, D.; Laman, H. Gsk3β and Tomm20 are substrates of the SCFFbxo7/PARK15 ubiquitin ligase associated with Parkinson’s disease. Biochem. J. 2016, 473, 3563–3580. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef]
- Geut, H.; Hepp, D.H.; Foncke, E.; Berendse, H.W.; Rozemuller, J.M.; Huitinga, I.; van de Berg, W.D.J. Neuropathological correlates of parkinsonian disorders in a large Dutch autopsy series. Acta Neuropathol. Commun. 2020, 8, 39. [Google Scholar] [CrossRef]
- Terracciano, C.; Nogalska, A.; Engel, W.K.; Askanas, V. In AbetaPP-overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: Effects mitigated by lithium and apparently relevant to sporadic inclusion-body myositis. J. Neurochem. 2010, 112, 389–396. [Google Scholar] [CrossRef]
- Brunello, C.A.; Merezhko, M.; Uronen, R.L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 04. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.R.; Cheng, K.C.; Chen, Y.R.; Lin, T.Y.; Cheung, C.H.A.; Wu, C.L.; Chiang, H.C. Dysfunction of different cellular degradation pathways contributes to specific β-amyloid42-induced pathologies. FASEB J. 2018, 32, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Rosen, K.M.; Moussa, C.E.; Lee, H.K.; Kumar, P.; Kitada, T.; Qin, G.; Fu, Q.; Querfurth, H.W. Parkin reverses intracellular beta-amyloid accumulation and its negative effects on proteasome function. J. Neurosci. Res. 2010, 88, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Herman, A.M.; Hoe, H.S.; Rebeck, G.W.; Moussa, C.E. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum. Mol. Genet. 2011, 20, 2091–2102. [Google Scholar] [CrossRef]
- Zhang, X.; Heng, X.; Li, T.; Li, L.; Yang, D.; Zhang, X.; Du, Y.; Doody, R.S.; Le, W. Long-term treatment with lithium alleviates memory deficits and reduces amyloid-βproduction in an aged Alzheimer’s disease transgenic mouse model. J. Alzheimers Dis. 2011, 24, 739–749. [Google Scholar] [CrossRef]
- Habib, A.; Sawmiller, D.; Li, S.; Xiang, Y.; Rongo, D.; Tian, J.; Hou, H.; Zeng, J.; Smith, A.; Fan, S.; et al. LISPRO mitigates β-amyloid and associated pathologies in Alzheimer’s mice. Cell Death Dis. 2017, 15, e2880. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Jimenez, S.; De Castro, V.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; Navarro, V.; Sanchez-Varo, R.; Sanchez-Mejias, E.; Davila, J.C.; et al. In vivo modification of Abeta plaque toxicity as a novel neuroprotective lithium-mediated therapy for Alzheimer’s disease pathology. Acta Neuropathol. Commun. 2013, 1, 73. [Google Scholar] [CrossRef]
- Shimada, K.; Motoi, Y.; Ishiguro, K.; Kambe, T.; Matsumoto, S.E.; Itaya, M.; Kunichika, M.; Mori, H.; Shinohara, A.; Chiba, M.; et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: Implications of autophagy promotion. Neurobiol. Dis. 2012, 46, 101–108. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Ryskalin, L.; Busceti, C.L.; Fornai, F. Molecular Mechanisms Linking ALS/FTD and Psychiatric Disorders, the Potential Effects of Lithium. Front. Cell. Neurosci. 2019, 13, 450. [Google Scholar] [CrossRef]
- Qiu, K.; Liang, W.; Wang, S.; Kong, T.; Wang, X.; Li, C.; Wang, Z.; Wu, Y. BACE2 degradation is mediated by both the proteasome and lysosome pathways. BMC Mol. Cell. Biol. 2020, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martín, T.; Cuchillo-Ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 2013, 34, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, H.; Nah, J.; Moon, S.; Lee, W.; Hong, S.H.; Kam, T.I.; Jung, Y.K. Essential role of POLDIP2 in Tau aggregation and neurotoxicity via autophagy/proteasome inhibition. Biochem. Biophys. Res. Commun. 2015, 462, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Guarascio, R.; Salih, D.; Yasvoina, M.; Edwards, F.A.; Cheetham, M.E.; van der Spuy, J. Negative Regulator of Ubiquitin-Like Protein 1 modulates the autophagy-lysosomal pathway via p62 to facilitate the extracellular release of tau following proteasome impairment. Hum. Mol. Genet. 2020, 29, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.J.; Lee, M.J. Dual Function of USP14 Deubiquitinase in Cellular Proteasomal Activity and Autophagic Flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, S.; Kim, E.; Lee, M.J. Negative-feedback coordination between proteasomal activity and autophagic flux. Autophagy 2019, 15, 726–728. [Google Scholar] [CrossRef]
- Srinivasan, V.; Bruelle, C.; Scifo, E.; Pham, D.D.; Soliymani, R.; Lalowski, M.; Lindholm, D. Dynamic Interaction of USP14 with the Chaperone HSC70 Mediates Crosstalk between the Proteasome, ER Signaling, and Autophagy. iScience 2020, 23, 100790. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef]
- Watabe, K.; Akiyama, K.; Kawakami, E.; Ishii, T.; Endo, K.; Yanagisawa, H.; Sango, K.; Tsukamoto, M. Adenoviral expression of TDP-43 and FUS genes and shRNAs for protein degradation pathways in rodent motoneurons in vitro and in vivo. Neuropathology 2014, 34, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Necchi, D.; Lomoio, S.; Scherini, E. Dysfunction of the ubiquitin-proteasome system in the cerebellum of aging Ts65Dn mice. Exp. Neurol. 2011, 232, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhang, S.; Tian, X.; Wu, C. Mask mitigates MAPT- and FUS-induced degeneration by enhancing autophagy through lysosomal acidification. Autophagy 2017, 13, 1924–1938. [Google Scholar] [CrossRef]
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456. [Google Scholar] [CrossRef]
- Onesto, E.; Rusmini, P.; Crippa, V.; Ferri, N.; Zito, A.; Galbiati, M.; Poletti, A. Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 266–280. [Google Scholar] [CrossRef]
- Crippa, V.; Galbiati, M.; Boncoraglio, A.; Rusmini, P.; Onesto, E.; Giorgetti, E.; Cristofani, R.; Zito, A.; Poletti, A. Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem. Soc. Trans. 2013, 41, 1598–1604. [Google Scholar] [CrossRef]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef]
- Guo, J.L.; Buist, A.; Soares, A.; Callaerts, K.; Calafate, S.; Stevenaert, F.; Daniels, J.P.; Zoll, B.E.; Crowe, A.; Brunden, K.R.; et al. The Dynamics and Turnover of Tau Aggregates in Cultured Cells: INSIGHTS INTO THERAPIES FOR TAUOPATHIES. J. Biol. Chem. 2016, 291, 13175–13193. [Google Scholar] [CrossRef]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 2017, 12, e0179375. [Google Scholar] [CrossRef]
- Martins, D.; English, A.M. SOD1 oxidation and formation of soluble aggregates in yeast: Relevance to sporadic ALS development. Redox Biol. 2014, 2, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Vicente, M.H.; Outeiro, T.F. The sour side of neurodegenerative disorders: The effects of protein glycation. J. Pathol. 2010, 221, 13–25. [Google Scholar]
- Natale, G.; Pompili, E.; Biagioni, F.; Paparelli, S.; Lenzi, P.; Fornai, F. Histochemical approaches to assess cell-to-cell transmission of misfolded proteins in neurodegenerative diseases. Eur. J. Histochem. 2013, 57, e5. [Google Scholar] [CrossRef] [PubMed]
- Grillo, M.A.; Colombatto, S. Advanced glycation end-products (AGEs): Involvement in aging and in neurodegenerative diseases. Amino Acids 2008, 35, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Nakashima, K.; Horiuchi, S.; Nagai, R.; Cleveland, D.W.; Liu, J.; Hirano, A.; Takikawa, M.; Kato, M.; Nakano, I.; et al. Formation of advanced glycation end-product-modified superoxide dismutase-1 (SOD1) is one of the mechanisms responsible for inclusions common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutation, and transgenic mice expressing human SOD1 gene mutation. Neuropathology 2001, 21, 67–81. [Google Scholar]
- Takamiya, R.; Takahashi, M.; Myint, T.; Park, Y.S.; Miyazawa, N.; Endo, T.; Fujiwara, N.; Sakiyama, H.; Misonou, Y.; Miyamoto, Y.; et al. Glycation proceeds faster in mutated Cu, Zn-superoxide dismutases related to familial amyotrophic lateral sclerosis. FASEB J. 2003, 17, 938–940. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Szegő, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Adrover, M.; Mariño, L.; Sanchis, P.; Pauwels, K.; Kraan, Y.; Lebrun, P.; Vilanova, B.; Muñoz, F.; Broersen, K.; Donoso, J. Mechanistic insights in glycation-induced protein aggregation. Biomacromolecules 2014, 15, 3449–3462. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ernst, L.; Grotzinger, N.; Hohn, A.; Breusing, N.; Reinheckel, T.; Grune, T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic. Res. 2010, 44, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Toki, S.; Chowei, H.; Saito, T.; Nakano, N.; Hayashi, Y.; Takeuchi, M.; Makita, Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 2001, 888, 256–262. [Google Scholar] [CrossRef]
- Maślińska, D.; Laure-Kamionowska, M.; Taraszewska, A.; Deręgowski, K.; Maśliński, S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011, 49, 295–300. [Google Scholar] [PubMed]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef]
- Gaczynska, M.; Rock, K.L.; Spies, T.; Goldberg, A.L. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc. Natl. Acad. Sci. USA 1994, 91, 9213–9217. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Ugras, S.; Daniels, M.J.; Fazelinia, H.; Gould, N.S.; Yocum, A.K.; Luk, K.C.; Luna, E.; Ding, H.; McKennan, C.; Seeholzer, S.; et al. Induction of the immunoproteasome subunit Lmp7 links proteostasis and immunity in α-synuclein aggregation disorders. EBioMedicine 2018, 31, 307–319. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T-cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Wagner, L.K.; Gilling, K.E.; Schormann, E.; Kloetzel, P.M.; Heppner, F.L.; Krüger, E.; Prokop, S. Immunoproteasome deficiency alters microglial cytokine response and improves cognitive deficits in Alzheimer’s disease-like APPPS1 mice. Acta Neuropathol. Commun. 2017, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Puttaparthi, K.; Elliott, J.L. Non-neuronal induction of immunoproteasome subunits in an ALS model: Possible mediation by cytokines. Exp. Neurol. 2005, 196, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; Bendotti, C. Major Histocompatibility Complex I Expression by Motor Neurons and Its Implication in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Heink, S.; Ludwig, D.; Kloetzel, P.M.; Krüger, E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef]
- Cebrián, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8, 114. [Google Scholar] [CrossRef]
- Leung, C.S. Endogenous Antigen Presentation of MHC Class II Epitopes through Non-Autophagic Pathways. Front. Immunol. 2015, 6, 464. [Google Scholar] [CrossRef]
- Pla, A.; Pascual, M.; Renau-Piqueras, J.; Guerri, C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain. Cell Death Dis. 2014, 5, e1066. [Google Scholar] [CrossRef]
- Zhang, H.M.; Fu, J.; Hamilton, R.; Diaz, V.; Zhang, Y. The mammalian target of rapamycin modulates the immunoproteasome system in the heart. J. Mol. Cell. Cardiol. 2015, 86, 158–167. [Google Scholar] [CrossRef]
- Lin, M.; Chandramani-Shivalingappa, P.; Jin, H.; Ghosh, A.; Anantharam, V.; Ali, S.; Kanthasamy, A.G.; Kanthasamy, A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 2012, 210, 308–332. [Google Scholar] [CrossRef]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.T.; Ureshino, R.P.; et al. Overexpression of α-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.M.; Wang, F.; Qi, D.; Liu, W.W.; Gu, C.; Mao, C.J.; Yang, Y.P.; Zhao, Z.; Hu, L.F.; Liu, C.F. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 2018, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, L.; Jiang, J.; Li, H.; Wu, Q.; Ooi, K.; Wang, J.; Feng, Y.; Zhu, D.; Xia, C. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J. Neuroinflamm. 2020, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, V.V.; Thandavarayan, R.A.; Arumugam, S.; Mizuno, M.; Nawa, H.; Suzuki, K.; Ko, K.M.; Krishnamurthy, P.; Watanabe, K.; Konishi, T. Schisandrin B Ameliorates ICV-Infused Amyloid β Induced Oxidative Stress and Neuronal Dysfunction through Inhibiting RAGE/NF-κB/MAPK and Up-Regulating HSP/Beclin Expression. PLoS ONE 2015, 10, e0142483. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Chen, W.; Wan, W.; Chen, Y.; Li, Y.; Zhang, C. Aβ(1-42) oligomer induces alteration of tight junction scaffold proteins via RAGE-mediated autophagy in bEnd.3 cells. Exp. Cell Res. 2018, 369, 266–274. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. Int. J. Mol. Sci. 2020, 21, 3028. https://doi.org/10.3390/ijms21083028
Limanaqi F, Biagioni F, Gambardella S, Familiari P, Frati A, Fornai F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. International Journal of Molecular Sciences. 2020; 21(8):3028. https://doi.org/10.3390/ijms21083028
Chicago/Turabian StyleLimanaqi, Fiona, Francesca Biagioni, Stefano Gambardella, Pietro Familiari, Alessandro Frati, and Francesco Fornai. 2020. "Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies" International Journal of Molecular Sciences 21, no. 8: 3028. https://doi.org/10.3390/ijms21083028
APA StyleLimanaqi, F., Biagioni, F., Gambardella, S., Familiari, P., Frati, A., & Fornai, F. (2020). Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. International Journal of Molecular Sciences, 21(8), 3028. https://doi.org/10.3390/ijms21083028