SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Body Composition and Biochemical Parameters
2.2. Albuminuria and Renal Function
2.3. Renal Hypertrophy, Glomerular and Podocyte Morphology
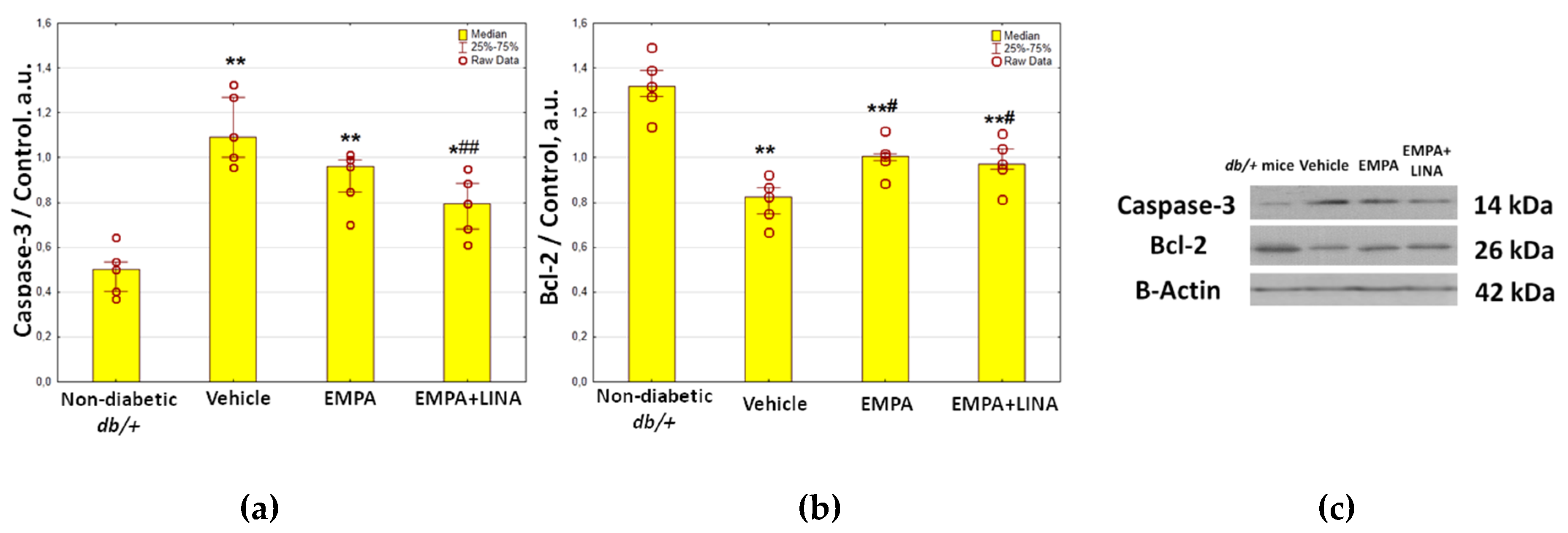
2.4. Expression of Autophagy and Apoptosis Regulators in the Renal Cortex
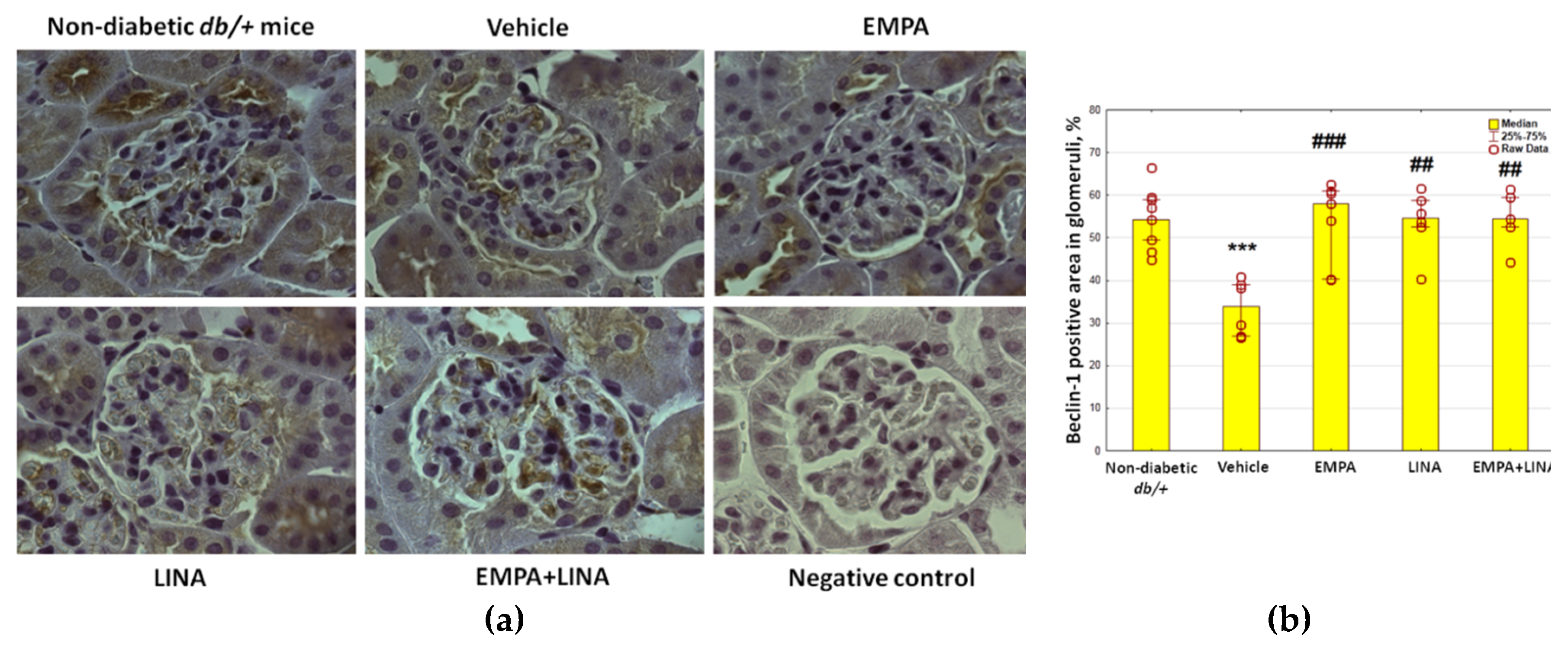
2.5. Glomerular Expression of the Autophagy Markers
2.6. Autophagy in Podocytes
2.7. Relationships between Autophagy Intensity, Glomerular Morphology and Biochemical Parameters
3. Discussion
3.1. Suppression of Renal Autophagy in Diabetes: Markers and Mechanisms
3.2. Renal Autophagy Suppression in Pathophysiology of Diabetic Kidney Disease
3.3. Reactivation of Autophagy: a Missing Link in the Mechanism of Renoprotection under SGLT2 and DPP4 Inhibition?
4. Materials and Methods
4.1. The Design of the Study
4.2. Ethical Issues
4.3. Laboratory Investigations
4.4. Light Microscopy and IHC
4.5. TEM
4.6. Westen Blotting
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| DPP4 | Dipeptidylpeptidase-4 |
| GLUT4 | Gucose transporter type 4 |
| IHC | Immunohistochemistry |
| mTOR | mammalian Target of rapamycin |
| NA | Numerical density |
| LAMP-1 | Lysosomal-associated membrane protein-1 |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PFs | Foot processes |
| SGLT2 | Sodium-glucose cotransporter-2 |
| TEM | Transmission electron microscopy |
| UACR | Urinary albumin-to-creatinine ratio |
| Vv | Volume density |
References
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic kidney disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Lin, J.S.; Susztak, K. Podocytes: The weakest link in diabetic kidney disease? Curr. Diab. Rep. 2016, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, S.; Zerbini, G. Glomerular endothelial cells versus podocytes as the cellular target in diabetic nephropathy. Acta Diabetol. 2018, 55, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Yasuda-Yamahara, M.; Kume, S.; Tagawa, A.; Maegawa, H.; Uzu, T. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy 2015, 11, 2385–2386. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, L.; Shi, Y.; Zhuang, S. Podocyte autophagy: A potential therapeutic target to prevent the progression of diabetic nephropathy. J. Diabetes Res. 2017, 2017, 3560238. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.A.; Wu, V.C.; Wang, C.Y. Autophagy in chronic kidney diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Yang, D.; Livingston, M.J.; Liu, Z.; Dong, G.; Zhang, M.; Chen, J.K.; Dong, Z. Autophagy in diabetic kidney disease: Regulation, pathological role and therapeutic potential. Cell Mol. Life Sci. 2018, 75, 669–688. [Google Scholar] [CrossRef]
- Zhang, Y.; Whaley-Connell, A.T.; Sowers, J.R.; Ren, J. Autophagy as an emerging target in cardiorenal metabolic disease: From pathophysiology to management. Pharmacol. Ther. 2018, 191, 1–22. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: The CARMELINA randomized clinical trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Bajaj, M.; Yang, H.C.; Ye, Y. Combined SGLT2 and DPP4 inhibition reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic nephropathy in mice with type 2 diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Zhou, B.; Xu, G. SGLT2 inhibitors: A novel choice for the combination therapy in diabetic kidney disease. Cardiovasc. Diabetol. 2017, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Fioretto, P.; Zambon, A.; Rossato, M.; Busetto, L.; Vettor, R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care 2016, 39 (Suppl. 2), S165–S171. [Google Scholar] [CrossRef]
- Korbut, A.I.; Klimontov, V.V. Empagliflozin: A new strategy for nephroprotection in diabetes. Diabetes Mellit. 2017, 20, 75–84. [Google Scholar] [CrossRef]
- Nespoux, J.; Vallon, V. SGLT2 inhibition and kidney protection. Clin. Sci. (Lond.) 2018, 132, 1329–1339. [Google Scholar] [CrossRef]
- Korbut, A.I.; Klimontov, V.V. Incretin-based therapy: Renal effects. Diabetes Mellit. 2016, 19, 53–63. [Google Scholar] [CrossRef]
- Scheen, A.J.; Delanaye, P. Renal outcomes with dipeptidyl peptidase-4 inhibitors. Diabetes Metab. 2018, 44, 101–111. [Google Scholar] [CrossRef]
- Gupta, S.; Sen, U. More than just an enzyme: Dipeptidyl peptidase-4 (DPP-4) and its association with diabetic kidney remodelling. Pharmacol. Res. 2019, 147, 104391. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Dong-Jin, K.; Seon, H.P.; Min, J.S.; Jaehee, K.; Jisu, O.; Dong, H.Y.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Renal Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Yang, C.C.; Chen, Y.T.; Wallace, C.G.; Chen, K.H.; Cheng, B.C.; Sung, P.H.; Li, Y.C.; Ko, S.F.; Chang, H.W.; Yip, H.K. Early administration of empagliflozin preserved heart function in cardiorenal syndrome in rat. Biomed. Pharmacother. 2019, 109, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Effects of newly introduced antidiabetic drugs on autophagy. Diabetes Metab. Syndr. 2019, 13, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Wang, S.S.; Han, Z.; Han, F.; Chang, Y.P.; Yang, Y.; Xue, M.; Sun, B.; Chen, L.M. Triptolide restores autophagy to alleviate diabetic renal fibrosis through the miR-141-3p/PTEN/Akt/mTOR Pathway. Mol. Ther. Nucleic Acids. 2017, 9, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Li, Z.; Xu, Y.; Yu, Y.; Zhou, Q.; Chen, L.; Wan, Q. Autophagy protects human podocytes from high glucose-induced injury by preventing insulin resistance. Metabolism 2016, 65, 1307–1315. [Google Scholar] [CrossRef]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Wang, Y.; Zeng, X. Apelin involved in progression of diabetic nephropathy by inhibiting autophagy in podocytes. Cell Death Dis. 2017, 8, e3006. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 2018, 152, 45–59. [Google Scholar] [CrossRef]
- Gong, J.; Zhan, H.; Li, Y.; Zhang, W.; Jin, J.; He, Q. Krüppel-like factor 4 ameliorates diabetic kidney disease by activating autophagy via the mTOR pathway. Mol. Med. Rep. 2019, 20, 3240–3248. [Google Scholar] [CrossRef]
- Lee, E.J.; Kang, M.K.; Kim, Y.H.; Kim, D.Y.; Oh, H.; Kim, S.I.; Oh, S.Y.; Kang, Y.H. Dietary chrysin suppresses formation of actin cytoskeleton and focal adhesion in AGE-exposed mesangial cells and diabetic kidney: Role of autophagy. Nutrients 2019, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zheng, H.; Huang, S.; You, N.; Xu, J.; Ye, X.; Zhu, Q.; Feng, Y.; You, Q.; Miao, H.; et al. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp. Cell Res. 2015, 337, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, B.; Li, H.; Ling, L.; Niu, J.; Gu, Y. Glucagon-like peptide-1 analog prevents obesity-related glomerulopathy by inhibiting excessive autophagy in podocytes. Am. J. Physiol. Renal Physiol. 2018, 314, F181–F189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, G.; Shen, H.; Gao, B.; Li, X.; Fu, J.; Zhou, J.; Ji, Q. Liraglutide inhibits autophagy and apoptosis induced by high glucose through GLP-1R in renal tubular epithelial cells. Int. J. Mol. Med. 2015, 35, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, S.; Xu, J.; Zhu, Q.; Ye, X.; Ding, D.; Yao, W.; Lu, Y. Dysregulation of lncRNAs GM5524 and GM15645 involved in high-glucose-induced podocyte apoptosis and autophagy in diabetic nephropathy. Mol. Med. Rep. 2018, 18, 3657–3664. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Lim, J.H.; Kim, H.W.; Kim, M.Y.; Kim, T.W.; Kim, E.N.; Kim, Y.; Chung, S.; Kim, Y.S.; Choi, B.S.; Kim, Y.S.; et al. Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis. 2018, 9, 270. [Google Scholar] [CrossRef]
- Inoki, K. mTOR signaling in autophagy regulation in the kidney. Semin. Nephrol. 2014, 34, 2–8. [Google Scholar] [CrossRef]
- De Rechter, S.; Decuypere, J.P.; Ivanova, E.; van den Heuvel, L.P.; De Smedt, H.; Levtchenko, E.; Mekahli, D. Autophagy in renal diseases. Pediatr. Nephrol. 2016, 31, 737–752. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Y. Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat. Rev. Nephrol. 2015, 11, 535–545. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Regulation of autophagy by TGF-β: Emerging role in kidney fibrosis. Semin. Nephrol. 2014, 34, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Liu, Q.; Liu, B. Research progress on mechanism of podocyte depletion in diabetic nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Regulating autophagy as a therapeutic target for diabetic nephropathy. Curr. Diab. Rep. 2017, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Interplay of adenosine monophosphate-activated protein kinase/sirtuin-1 activation and sodium influx inhibition mediates the renal benefits of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes: A novel conceptual framework. Diabetes Obes. Metab. 2020, 22, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, Y.; Tan, X.; Zhang, L.; Zhang, H.; Li, Z.; Liu, S.; Li, R.; Lin, T.; Liao, R.; et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J. Pathol. 2018, 245, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yang, S.; Zhu, X.; Sun, D.; Sun, D.; Jiang, X.; Zhang, C.; Wang, L. The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells. Cell Signal. 2019, 62, 109334. [Google Scholar] [CrossRef]
- Malik, S.A.; Mariño, G.; BenYounès, A.; Shen, S.; Harper, F.; Maiuri, M.C.; Kroemer, G. Neuroendocrine regulation of autophagy by leptin. Cell Cycle 2011, 10, 2917–2923. [Google Scholar] [CrossRef]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef]
- Audzeyenka, I.; Rogacka, D.; Piwkowska, A.; Angielski, S.; Jankowski, M. Viability of primary cultured podocytes is associated with extracellular high glucose-dependent autophagy downregulation. Mol. Cell Biochem. 2017, 430, 11–19. [Google Scholar] [CrossRef]
- Coward, R.; Fornoni, A. Insulin signaling: Implications for podocyte biology in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 104–110. [Google Scholar] [CrossRef]
- Guzman, J.; Jauregui, A.N.; Merscher-Gomez, S.; Maiguel, D.; Muresan, C.; Mitrofanova, A.; Diez-Sampedro, A.; Szust, J.; Yoo, T.H.; Villarreal, R.; et al. Podocyte-specific GLUT4-deficient mice have fewer and larger podocytes and are protected from diabetic nephropathy. Diabetes 2014, 63, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Ezquerro, S.; Frühbeck, G.; Rodríguez, A. Ghrelin and autophagy. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 402–408. [Google Scholar] [CrossRef]
- Wang, Z.H.; Ren, W.Y.; Zhu, L.; Hu, L.J. Plasminogen activator inhibitor-1 regulates LPS induced inflammation in rat macrophages through autophagy activation. Sci. World J. 2014, 2014, 189168. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Chuang, H.C.; Lee, Y.H.; Lin, Y.F.; Chiu, Y.J.; Wang, Y.L.; Wu, M.S.; Chiu, H.W. Induction of fibrosis and autophagy in kidney cells by vinyl chloride. Cells 2019, 8, 601. [Google Scholar] [CrossRef] [PubMed]
- Kanasaki, K.; Kawakita, E.; Koya, D. Relevance of Autophagy Induction by Gastrointestinal Hormones: Focus on the Incretin-Based Drug Target and Glucagon. Front. Pharmacol. 2019, 10, 476. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A.; Shah, S.V. Autophagy function and regulation in kidney disease. Biomolecules 2020, 10, 100. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, H.; Man, F.; Guo, Z.; Xu, J.; Yan, W.; Li, J.; Pan, Q.; Wang, W. Sitagliptin protects cardiac function by reducing nitroxidative stress and promoting autophagy in Zucker diabetic fatty (ZDF) rats. Cardiovasc. Drugs Ther. 2018, 32, 541–552. [Google Scholar] [CrossRef]
- Murase, H.; Kuno, A.; Miki, T.; Tanno, M.; Yano, T.; Kouzu, H.; Ishikawa, S.; Tobisawa, T.; Ogasawara, M.; Nishizawa, K.; et al. Inhibition of DPP-4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats. Cardiovasc. Diabetol. 2015, 14, 103. [Google Scholar] [CrossRef]
- Gu, Y.; Ma, C.T.; Gu, H.L.; Shi, L.; Tian, X.T.; Xu, W.Q. Sitagliptin improves cardiac function after myocardial infarction through activation of autophagy in streptozotocin-induced diabetic mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8973–8983. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhou, J.; Song, S.; Kong, W.; Xia, W.; Chen, L.; Zeng, T. Dipeptidyl-peptidase 4 inhibitor sitagliptin ameliorates hepatic insulin resistance by modulating inflammation and autophagy in ob/ob mice. Int. J. Endocrinol. 2018, 2018, 8309723. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi Jigheh, Z.; Ghorbani Haghjo, A.; Argani, H.; Roshangar, L.; Rashtchizadeh, N.; Sanajou, D.; Nazari Soltan Ahmad, S.; Rashedi, J.; Dastmalchi, S.; Mesgari Abbasi, M. Empagliflozin alleviates renal inflammation and oxidative stress in streptozotocin-induced diabetic rats partly by repressing HMGB1-TLR4 receptor axis. Iran. J. Basic Med. Sci. 2019, 22, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Sharkovska, Y.; Reichetzeder, C.; Alter, M.; Tsuprykov, O.; Bachmann, S.; Secher, T.; Klein, T.; Hocher, B. Blood pressure and glucose independent renoprotective effects of dipeptidyl peptidase-4 inhibition in a mouse model of type-2 diabetic nephropathy. J. Hypertens. 2014, 32, 2211–2223. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova, Y.S.; Bgatova, N.P.; Klimontov, V.V.; Ischenko, I.Y.; Michurina, S.V.; Myakina, N.E.; Zavyalov, E.L. Effect of linagliptin on structural changes in the kidney in experimental type 2 diabetes mellitus. Bull. Exp. Biol. Med. 2016, 161, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Mandarim-de-Lacerda, C.A.; Del Sol, M. Tips for studies with quantitative morphology (morphometry and stereology). Int. J. Morphol. 2017, 35, 1482–1494. [Google Scholar] [CrossRef]
- Eskelinen, E.L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008, 4, 257–260. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Higgins, G.C.; Nguyen, T.V.; Ramm, G.; Coughlan, M.T. Methods in renal research: Measurement of autophagic flux in the renal cortex ex vivo. Nephrology (Carlton) 2018, 23, 815–820. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; Freeman, W.H., Ed.; Organelles of the Eukaryotic Cell: New York, NY, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK21743/ (accessed on 10 April 2020).
- Cheville, N.F.; Stasko, J. Techniques in electron microscopy of animal tissue. Vet. Pathol. 2014, 51, 28–41. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Week of Age | db/+ Mice (Non-Diabetic Control) | db/db Mice | |||
|---|---|---|---|---|---|---|
| Vehicle | EMPA | LINA | EMPA + LINA | |||
| n | 9 | 10 | 9 | 6 | 6 | |
| Body weight, g | 8 | 26.8 (25.0–31.1) | 37.1 (32.1–41.6)*** | 37.4 (28.5–41.5)*** | 36.5 (29.6–40.3)** | 37.3 (35.9–39.5)*** |
| 16 | 24.8 (21.6–27.7)+ | 37.8 (34.3–40.4)*** | 52.6 (42.5–62.2)***##++ | 45.3 (39.5–47.5)***§§+ | 50.4 (46.9–52.3)***#ØØ+ | |
| Fat mass, g | 8 | 3.1 (1.9–4.1) | 18.3 (16.2–22.5)*** | 18.4 (13.5–20.6)*** | 18.0 (15.1–19.5)*** | 18.1 (16.7–19.5)*** |
| 16 | 2.7 (2.1–4.5) | 19.6 (17.4–22.7)*** | 28.0 (22.2–35.3)***###++ | 23.4 (20.3–26.4)***##+ | 26.7 (23.2–28.3)***###+ | |
| Lean mass, g | 8 | 22.2 (21.0–24.8) | 17.1 (16.3–18.7)*** | 18.2 (13.4–19.8)*** | 16.0 (13.8–17.2)*** | 18.5 (15.8–19.1)*** |
| 16 | 21.2 (19.1–22.5)+ | 16.1 (14.3–22.4)** | 21.4 (19.1–24.4)++ | 17.6 (14.9–20.7)**§+ | 20.6 (19.5–21.5)Ø + | |
| Total water, g | 8 | 19.5 (18.4–21.5) | 14.8 (14.5–16.4)*** | 15.8 (11.6–17.6)*** | 12.5 (9.6–14.1)*** | 16.0 (13.7–16.5)*** |
| 16 | 18.4 (16.5–19.4)+ | 14.1 (12.3–19.2)* | 18.8 (16.7–21.4)++ | 15.5 (13.0–18.1)**§+ | 18.2 (17.3–18.7)Ø + | |
| Parameter | Week of Age | db/+ Mice (Non-Diabetic Control) | db/db Mice | |||
|---|---|---|---|---|---|---|
| Vehicle | EMPA | LINA | EMPA + LINA | |||
| n | 9 | 10 | 9 | 6 | 6 | |
| Glucose, mmol/L | 8 | 9.5 (5.1–11.4) | 28.5 (16.8–40.1)*** | 23.1 (15.3–35.4)*** | 27.5 (20.6–48.8)*** | 25.9 (20.4–33.0)*** |
| 16 | 9.5 (8.5–12.2) | 32.7 (22.5–53.1) *** | 16.1 (9.9–23.6)***###+ | 29.9 (25.8–34.6)***§§ | 15.2 (11.7–23.5)***ØØ##+ | |
| Fructosamine, μmol/L | 8 | 237 (217–249) | 456 (424–511)*** | 480 (425–579)*** | 436 (383–487)*** | 430 (406; 475)*** |
| 16 | 239 (222–296) | 622 (524–672)***++ | 468 (341–491)**+ | 650 (591–691)***§§+ | 380 (342–392)*ØØ+ | |
| Glycated albumin, μmol/L | 8 | 107 (103–127) | 227 (206–239)*** | 235 (217–261)*** | 209 (156–256)*** | 217 (203–228)*** |
| 16 | 117 (109–133) | 283 (252–349)***++ | 210 (166–245)**##+ | 315 (225–376)***§§ | 178 (157–182)*###ØØ+ | |
| Leptin, ng/mL | 8 | 3.30 (1.40–6.54) | 97.1 (53.2–114.4)*** | 93.4 (80.8–133.1)*** | 92.9 (76.0–118.4)*** | 89.9 (66.6–119.1)*** |
| 16 | 3.80 (1.50–6.30) | 90.0 (21.2–151.4)*** | 136.4 (53.0–171.2)***#+ | 96.5 (51.6–172.6)*** | 139.1 (83.7–200.6)*** | |
| Insulin, ng/mL | 8 | 5.59 (3.43–18.0) | 25.6 (11.5–45.2)*** | 21.2 (10.8–40.8)** | 22.7 (15.3–54.0)** | 22.1 (13.8–76.7)*** |
| 16 | 10.0 (2.10–22.2) | 22.1 (9.8–33.2)** | 20.8 (5.95–56.7)* | 13.9 (7.2–23.7) | 33.3 (13.6–72.9)**Ø | |
| Glucagon, ng/mL | 8 | 370 (250–2670) | 660 (190–2760) | 450 (290–2180) | 375 (260–2090) | 300 (170–780) |
| 16 | 370 (150–2120) | 605 (260–2960) | 390 (240–860) | 325 (190–810) | 310 (200–460)# | |
| Ghrelin, ng/mL | 8 | 0.99 (0.24–4.27) | 0.42 (0.10–1.57)** | 0.50 (0.25–1.15)** | 0.37 (0.25–0.82)** | 0.39 (0.32–1.13)** |
| 16 | 1.47 (0.76–6.31) | 1.27 (0.57–2.26)* | 0.81 (0.28–2.58) | 0.45 (0.20–1.14)***§ | 0.77 (0.08–1.47)* | |
| PAI-1, ng/mL | 8 | 1.52 (0.70–3.24) | 2.76 (1.41–4.23)* | 2.07 (1.00–6.01) | 1.84 (0.48–4.20) | 2.25 (1.72–3.91)* |
| 16 | 3.22 (0.40–4.36) | 1.64 (0.09–3.76) | 2.22 (0.79–5.12) | 1.85 (0.63–4.17) | 2.82 (0.98–6.05) | |
| Parameter | Week of Age | db/+ Mice (Non-Diabetic Control) | db/db Mice | |||
|---|---|---|---|---|---|---|
| Vehicle | EMPA | LINA | EMPA + LINA | |||
| n | 9 | 10 | 9 | 6 | 6 | |
| UACR, mg/mmol | 8 | 1.50 (0.60–4.00) | 14.8 (10.0–26.9)*** | 16.3 (12.8–30.0)*** | 26.8 (19.4–54.7)*** | 23.5 (14.3–41.4)*** |
| 16 | 1.80 (0.10–3.10) | 21.4 (14.4–29.4)*** | 11.4 (4.00–22.7)***###++ | 6.9 (4.40–22.4)***##+ | 10.6 (4.50–17.6)***##+ | |
| Plasma creatinine, μmol/L | 8 | 67.0 (47.0–78.0) | 76.4 (70.2–84.2) | 70.5 (54.0–82.2) | 70.3 (52.8–103.5) | 70.4 (66.6– 82.2) |
| 16 | 61.8 (54.0–71.7) | 75.6 (63.9–89.4)** | 81.3 (55.8–91.2)* | 71.7 (67.8–99.0)** | 80.4 (65.7–91.2)** | |
| Parameter | db/+ Mice (Non-Diabetic Control) | db/db Mice | |||
|---|---|---|---|---|---|
| Vehicle | EMPA | LINA | EMPA + LINA | ||
| n | 9 | 10 | 9 | 6 | 6 |
| Mesangial fractional volume, % | 14.4 (9.8–18.5) | 38.6 (34.5–42.7)*** | 25.5 (20.4–35.8)**### | 31.1 (27.3–34.6)**## | 28.5 (26.1–30.0)**# |
| GBM width, nm | 135 (116–157) | 163 (135–203)* | 139 (116–225)# | 150 (126–165)# | 146 (108–152)# |
| Width of podocyte FPs, nm | 220 (191–242) | 372 (299–426)** | 203 (162–317)## | 189 (172–243)# | 202 (165–252)## |
| NA of podocyte FPs, nm–1 | 3.39 (3.00–3.79) | 2.73 (1.92–3.51)* | 3.31 (2.32–4.18)# | 3.33 (2.85–4.07)# | 3.25 (2.58–3.56)# |
| Parameter | db/+ Mice (Non-Diabetic Control) | db/db Mice Groups | |||
|---|---|---|---|---|---|
| Vehicle | EMPA | LINA | EMPA + LINA | ||
| n | 5 | 6 | 5 | 5 | 5 |
| Autophagosomes, Vv, % | 2.10 (1.80–2.86) | 0.80 (0.61–1.68)*** | 2.09 (1.65–2.5)### | 2.23 (1.73–2.35)### | 2.14 (1.57–2.31)### |
| Autolysosomes, Vv, % | 2.37 (1.85–3.21) | 1.09 (0.84–1.69)*** | 2.25 (1.37–3.09)### | 2.37 (1.58–2.96)### | 2.22 (1.58–3.04)### |
| Lysosomes, Vv, % | 2.94 (2.42–4.00) | 2.22 (1.22–2.92)*** | 3.16 (2.06–3.83)## | 2.64 (2.08–3.52)# | 2.89 (2.44–3.58)### |
| Parameter | Autophagosomes, Vv | Autolysosomes, Vv | Lysosomes, Vv |
|---|---|---|---|
| Glomerular beclin-1 (IHC) | r = 0.58 p = 0.0001, n = 26 | r = 0.48 p = 0.002, n = 26 | r = 0.25 p > 0.05, n = 26 |
| Glomerular LAMP-1 (IHC) | r = 0.46 p = 0.003, n = 26 | r = 0.50 p = 0.001, n = 26 | r = 0.42 p = 0.008, n = 26 |
| Renal LC3B (WB) | r = −0.57 p = 0.009, n = 20 | r = −0.69 p = 0.0008, n = 20 | r = −0.55 p = 0.01, n = 20 |
| Renal LAMP-1 (WB) | r = 0.50 p = 0.02, n = 20 | r = 0.55 p = 0.01, n = 20 | r = 0.63 p = 0.003, n = 20 |
| Renal caspase-3 (WB) | r = −0.35 p > 0.05, n = 20 | r = −0.56 p = 0.01, n = 20 | r = −0.44 p > 0.05, n = 20 |
| Renal Bcl-2 (WB) | r = 0.54 p = 0.01, n = 20 | r = 0.57 p = 0.009, n = 20 | r = 0.63 p = 0.003, n = 20 |
| Parameter | Glomerular beclin-1, % | Glomerular LAMP-1, % | Autophagosomes in Podocytes, Vv | Autolysosomes in Podocytes, Vv | Lysosomes in Podocytes, Vv |
|---|---|---|---|---|---|
| Kidneys weight/lean mass ratio | r = −0.13 p > 0.05, n = 33 | r = −0.19 p > 0.05, n = 33 | r = −0.31 p > 0.05, n = 26 | r = −0.43 p = 0.007, n = 26 | r = −0.45 p = 0.007, n = 26 |
| Mesangial fractional volume | r = −0.39 p = 0.02, n = 40 | r = −0.37 p = 0.03, n = 40 | r = −0.36 p = 0.03, n = 40 | r = −0.50 p = 0.002, n = 40 | r = −0.32 p = 0.009, n = 40 |
| Mean width of GBM | r = −0.41 p = 0.03, n = 26 | r = −0.40 p = 0.03, n = 26 | r = −0.54 p = 0.003, n = 26 | r = −0.45 p = 0.01, n = 26 | r = −0.26 p > 0.05, n = 26 |
| Mean width of podocyte FPs | r = −0.62 p = 0.0004, n = 26 | r = −0.29 p > 0.05, n = 26 | r = −0.58 p = 0.0009, n = 26 | r = −0.49 p = 0.007, n = 26 | r = −0.41 p = 0.03, n = 26 |
| NA of podocyte FPs | r = 0.27 p > 0.05, n = 26 | r = 0.32 p > 0.05, n = 26 | r = 0.54 p = 0.003, n = 26 | r = 0.47 p = 0.01, n = 26 | r = 0.35 p > 0.05, n = 26 |
| Parameter | Renal LC3B | Renal LAMP-1 | Renal Caspase-3 | Renal Bcl-2 |
|---|---|---|---|---|
| Kidney weight/lean mass ratio | r = 0.40 p > 0.05 | r = −0.60 p = 0.005 | r = 0.64 p = 0.002 | r = −0.64 p = 0.002 |
| Mesangial fractional volume | r = 0.45 p = 0.045 | r = −0.71 p = 0.0004 | r = 0.67 p = 0.001 | r = −0.71 p = 0.0004 |
| Mean width of GBM | r = 0.67 p = 0.002 | r = −0.45 p = 0.045 | r = 0.69 p = 0.002 | r = −0.53 p = 0.02 |
| Mean width of podocyte FPs | r = 0.56 p = 0.02 | r = −0.38 p > 0.05 | r = 0.52 p = 0.02 | r = −0.44 p = 0.048 |
| NA of podocyte FPs | r = −0.46 p = 0.04 | r = 0.38 p > 0.05 | r = −0.62 p = 0.004 | r = 0.46 p = 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. https://doi.org/10.3390/ijms21082987
Korbut AI, Taskaeva IS, Bgatova NP, Muraleva NA, Orlov NB, Dashkin MV, Khotskina AS, Zavyalov EL, Konenkov VI, Klein T, et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. International Journal of Molecular Sciences. 2020; 21(8):2987. https://doi.org/10.3390/ijms21082987
Chicago/Turabian StyleKorbut, Anton I., Iuliia S. Taskaeva, Nataliya P. Bgatova, Natalia A. Muraleva, Nikolai B. Orlov, Maksim V. Dashkin, Anna S. Khotskina, Evgenii L. Zavyalov, Vladimir I. Konenkov, Thomas Klein, and et al. 2020. "SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes" International Journal of Molecular Sciences 21, no. 8: 2987. https://doi.org/10.3390/ijms21082987
APA StyleKorbut, A. I., Taskaeva, I. S., Bgatova, N. P., Muraleva, N. A., Orlov, N. B., Dashkin, M. V., Khotskina, A. S., Zavyalov, E. L., Konenkov, V. I., Klein, T., & Klimontov, V. V. (2020). SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. International Journal of Molecular Sciences, 21(8), 2987. https://doi.org/10.3390/ijms21082987