Abstract
A perennial task is to prevent the occurrence and/or recurrence of most frequent or life-threatening cardiac arrhythmias such as atrial fibrillation (AF) and ventricular fibrillation (VF). VF may be lethal in cases without an implantable cardioverter defibrillator or with failure of this device. Incidences of AF, even the asymptomatic ones, jeopardize the patient’s life due to its complication, notably the high risk of embolic stroke. Therefore, there has been a growing interest in subclinical AF screening and searching for novel electrophysiological and molecular markers. Considering the worldwide increase in cases of thyroid dysfunction and diseases, including thyroid carcinoma, we aimed to explore the implication of thyroid hormones in pro-arrhythmic signaling in the pathophysiological setting. The present review provides updated information about the impact of altered thyroid status on both the occurrence and recurrence of cardiac arrhythmias, predominantly AF. Moreover, it emphasizes the importance of both thyroid status monitoring and AF screening in the general population, as well as in patients with thyroid dysfunction and malignancies. Real-world data on early AF identification in relation to thyroid function are scarce. Even though symptomatic AF is rare in patients with thyroid malignancies, who are under thyroid suppressive therapy, clinicians should be aware of potential interaction with asymptomatic AF. It may prevent adverse consequences and improve the quality of life. This issue may be challenging for an updated registry of AF in clinical practice. Thyroid hormones should be considered a biomarker for cardiac arrhythmias screening and their tailored management because of their multifaceted cellular actions.
1. Introduction
Atrial fibrillation (AF) and ventricular fibrillation (VF) are clinically relevant and potentially life-threatening arrhythmias. Despite some differences, both AF and VF have been assumed to occur due to abnormalities in the electrical activity involved in impulse initiation and impulse propagation [,,,,,,,,]. The former is due to an enhanced automaticity of the cardiomyocytes (i.e., pacemaker-like activity) or triggered activity expressed as early after-depolarization (EAD) or delayed after-depolarization (DAD). The latter is associated with a conduction block promoting a re-entrant excitation (most likely due to an electrical uncoupling), disorders at the intercellular connexin (Cx) channels, myocardial structural remodeling (hypertrophy and/or fibrosis, adiposity), and variations in the refractory periods. As mentioned in this review and depicted in Figure 1, thyroid hormones (TH) can be involved in all of these mechanisms, as well as in the modulation of the autonomous nervous system (ANS) and the renin–angiotensin–aldosterone system (RAAS) that affect arrhythmogenesis.
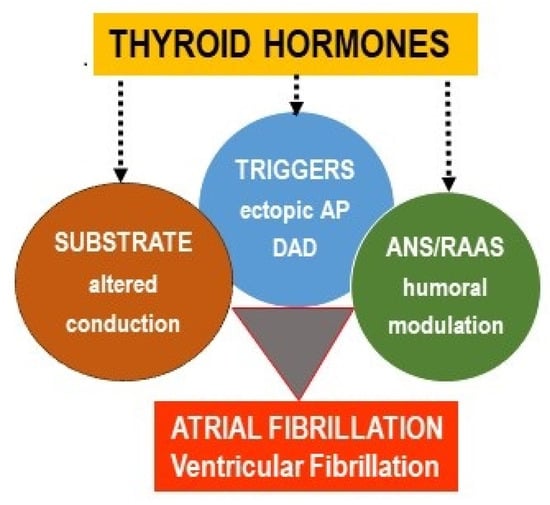
Figure 1.
An excess of thyroid hormones (TH) promotes the occurrence of the main factors involved in cardiac arrhythmogenesis: Substrate, triggers, autonomous nervous system (ANS), and renin–angiotensin–aldosterone system (RAAS) imbalance.
The thyroid gland produces thyroxine (T4), which is a relatively inactive prohormone, and lower amounts of the active hormone, triiodothyronine (T3). About 20% of T3 is made by the thyroid gland and the other 80% comes from T4 upon conversion by type 1 iodothyronine deiodinases (IDs), which is widely distributed. Type 2 ID catalyzes the inactivation of FT4 and FT3 while type 3 ID acts like type 1 ID, and is expressed in skeletal and cardiac muscles, thus providing local intracellular production of T3 [].
The release of TH from the thyroid gland is controlled by the thyrotrophin-releasing hormone from the hypothalamus in the brain and by the thyroid-stimulating hormone (TSH) produced by the pituitary gland. This forms part of a feedback loop known as the hypothalamic–pituitary–thyroid axis []. The thyroid gland can become overactive in hyperthyroidism or underactive in hypothyroidism. The latter is often accompanied by an enlargement of the thyroid gland known as goiter. Thyrotoxicosis may be the result of hyperthyroidism as in Graves’ disease (autoimmune hyperthyroidism), inflammation of the thyroid, benign thyroid tumor, or due to factors that affect hypothalamic, pituitary, or thyroid function []. Hyperthyroidism can also be caused by an acquired immune deficiency syndrome along with anti-retroviral therapy []. Moreover, a not-so-infrequent cause of hyperthyroidism in clinical practice is an iatrogenic one due to the adverse action of drugs, namely amiodarone. Apart from that, various gastrointestinal disorders may impede the absorption of T4 and increase the risk of developing iatrogenic hyperthyroidism [].
Both overt and subclinical (latent) hyperthyroidism are endocrine disorders that occur as a result of excessive TH secretion. TSH is a sensitive indicator of thyroid function. Subclinical hyperthyroidism is common in the general population and its frequency is variable, depending on age, sex, and iodine status [,,], and has been defined as lower serum TSH levels (<0.10 mlU/L), while the free T4 and T3 concentrations are within the reference range. The euthyroid status is defined as the condition wherein the TSH level is within the range from 0.45 to 4.49 mlU/L. In the National Health and Nutrition Examination Survey, 2.5% of the population had a serum TSH below the lower limit of the reference range [].
TH affect heart metabolism, electrical properties, and function through the interplay of genomic and non-genomic mechanisms of action [,,,,]. Accordingly, chronic and acute changes in the circulating TH have a fundamental impact on cardiac electrophysiology, Ca2+ handling, and structural remodeling. Disorders of these cellular factors due to a TH imbalance affect the susceptibility of the heart to arrhythmias [,,].
Both overt and subclinical hyperthyroidism in humans increases the risk for cardiac arrhythmias, especially AF [,,]. Recent data indicate that even in euthyroid individuals, the high-normal range of the circulating free T4 is associated with an increased AF incidence []. The incidence of VF attributed solely to the thyroid status imbalance is less frequent in humans, unlike the experimental animals, which are prone to both AF and VF in response to an excess of TH [,,].
It should be noted that an exogenous T4 administration results in similar increases in circulation, as well as the T3 levels, in cardiac tissues []. Evidence suggests that thyroid dysfunction and thyroid diseases associated with a TH suppressive therapy may exert a clinically relevant impact on the susceptibility of the heart to arrhythmias, as well as the outcomes of treatment. This issue is even more complicated due to the fact that TH biosynthesis machinery has been detected in the heart and has been altered due to cardiac pathology [].
2. Atrial Fibrillation and Pro-Arrhythmic Signaling of TH
2.1. A Short Overview on AF
Concerning AF, the cardiomyocyte sleeves overlapping the pulmonary veins along with Ca2+ handling disorders are the sources of the ectopic electrical activity, while re-entry circuits are promoted by the atrial tissue heterogeneity and disorders in the intercellular electrical coupling mediated by connexin (Cx) channels [,,,,,,,,]. The association of AF with the atrial Cx37 and Cx40 gene polymorphisms [], as well as somatic mutations in GJA5 (encoding Cx40), have been identified in AF [,]. As a pulmonary veins isolation-based approach can resolve AF in 50%–70% of patients, it implies that other drivers of AF remain to be determined []. TH may be one of those drivers for AF.
Risk factors for AF, such as aging, obstructive sleep apnea, diabetes, hypertension, dyslipidemia, obesity, cancer, renal dysfunction, and thyroid diseases, which are all accompanied by deleterious oxidative stress, may act synergistically to cause AF [,,,,,,,], whereby the noncoding microRNAs translate cellular stressors, such as reactive oxygen species, into AF pathogenesis []. Emerging evidence suggests a significant role of the altered atrial metabolism, phosphorylation of proteins, inflammatory and autoimmune channelopathies, and presence of autoantibodies to the M2-muscarinic and β 1-adrenergic receptors in the pathogenesis of AF [,,,,,,,,]. Due to these mentioned chronic stressors implicated in electrical remodeling and poor risk factors control, the incidence of AF increases globally.
AF, as recognized according to an irregular R–R interval and a missing P wave in an ECG, is a highly prevalent arrhythmia promoting heart failure, embolic stroke, and death []. Even short, subclinical episodes of AF are associated with an increased risk of stroke []. Paroxysmal, as well as sustained and permanent forms of AF, confer a significant clinical burden and worsens the patient’s quality of life.
Management of AF includes antiarrhythmic drug therapies that are often ineffective in terminating AF or preventing its recurrences, possibly because these drugs target a single pathophysiological mechanism []. Catheter ablation of the arrhythmogenic triggers, another option of AF treatment, does not prevent recurrence of AF, probably because of the persistence of the arrhythmogenic substrate [,]. In the advanced form of AF, the abnormal atrial substrate, Cx43, Cx40, and Cx45 abnormalities are thought to act as drivers of arrhythmia perpetuation [,,,]. Modulation of the autonomic nervous system has shown promising alternatives to the standard AF treatment []. Nevertheless, a better understanding of the modifiable biomarkers, including an altered thyroid status, and molecular factors, including autoantibodies, may provide us with a chance to prevent AF or to tailor the treatment to avoid harmful consequences. It is noteworthy that women have worse and often atypical symptoms, as well as a higher risk for stroke and death, associated with AF compared to the risk in men [].
It should, however, be emphasized that a considerable number of individuals have no AF symptoms [,], which is a major difficulty in an arrhythmia screening for detection of AF. Therefore, silent or subclinical AF is a major health concern, particularly because of its association with stroke. There is a need for novel approaches, as well as diagnostic and prognostic biomarkers []. Intermittent hand-held ECG recording revealed that the prevalence of AF in a population-based study was about 30% []. Patients with AF exhibit increased levels of the circulating N-terminal B-type natriuretic peptide (NT-proBNP), as well as the fibroblast growth factor-23 (FGF-23). Elevation of these markers can predict the development of AF in high-risk subjects or identify patients with AF [,,]. In this context, it appears relevant to monitor TH status as well.
2.2. Thyroid Status Imbalance Promoting AF
TH are one of the factors associated with AF and potential drivers of AF [,,]. Increased automaticity and an enhanced triggered activity may increase the arrhythmogenic activity of cardiomyocytes at the pulmonary veins in hyperthyroidism []. The propensity for AF is increased in animals [], as well as in humans with overt or subclinical hyperthyroidism that is more common in the general population and often accompanies various diseases [,,]. Indeed, TSH levels ≤0.1 mIU/L have been linked with a three-fold increase in AF risk []. Even in euthyroid subjects with normal serum TSH levels, the free T4 concentration has been independently associated with AF []. Maximum P-wave duration and P-wave dispersion (indicators for the risk of paroxysmal AF) were longer in patients with endogenous and exogenous subclinical hyperthyroidism []. The incidence of paroxysmal AF is higher in toxic nodular goiter [] and in patients suffering from Graves’ disease [,], which is caused by the presence of TSH autoantibodies directed toward the G protein-coupled TSH receptor []. The rate of intra- and extra-thyroidal conversion of T4 to T3 is elevated in Graves’ disease mostly due to an increased deiodase-1 activity. Thyroidectomy in Graves’ disease has been reported to abolish the AF load []. It is worthwhile to note that the occurrence of AF in this condition is associated with concomitant autoantibodies toward the β1-adrenergic and the M2 muscarinic receptors [,,,,]. These autoantibodies and T4 facilitated the induction of AF in animal models as well []. Autoantibodies activating the G protein-coupled β1-adrenergic receptors in the heart have been implicated in the development of both AF and VF [,,,]. Thus, autonomic autoantibodies along with TH potentiate the vulnerability of the heart to AF. Considering the impact of TH on the development of AF, the screening for AF is highly relevant not only in patients with an overt but particularly with a subclinical hyperthyroidism.
Moreover, TH affect the outcomes after an invasive treatment of AF. Accordingly, high circulating T3, as well as high-normal T4 or lower TSH levels, have been associated with AF recurrence after an arrhythmogenic foci ablation [,,,,]. Free T4 levels influence the success rate of ablation procedures even in the normal range. Likewise, TH replacement therapy exerts an impact on the AF ablation outcome []. Despite sophisticated invasive approaches, the recurrence of AF remains an unresolved problem in clinical practice, challenging for further research [].
3. Impact of Thyroid Hormones on Ventricular Arrhythmias
3.1. A Brief Overview on VF
Concerning VF, development of this life-threatening arrhythmia is similar to the development of a multifactorial AF [,,]. VF is triggered by the dysfunction of ion and connexin channels along with abnormal Ca2+-handling and facilitated in the presence of an arrhythmogenic structural substrate (such as myocardial hypertrophy, fibrosis, and misdistribution of connexins). All these events are influenced by the modulating factors, such an ischemia or autonomous nervous system (ANS) and hormonal imbalance, including TH. Specific QRS complex patterns, recognized due to hypertrophy, may potentially predict ventricular arrhythmias []. When structural abnormalities are not evident, autoimmune channelopathies have been established as a novel mechanism in cardiac arrhythmias [,,,,,]. In particular, proarrhythmic autoantibodies targeting calcium, potassium, or sodium channels and anti-desmosome antibodies in the heart have been identified. These autoantibodies promote conduction disturbances and induce substantial electrophysiological changes facilitating life-threatening ventricular arrhythmias.
Despite recognizing the basic mechanisms that can cause VF, the changes in the cardiac electrical properties remain poorly understood. Electrical disturbances result from the immediate operation of one or other arrhythmogenic mechanisms in different heart conditions. Accordingly, higher levels of total T3 have been positively associated with the heart rate, QTc, and negatively associated with the PR interval and QRS duration [].
Notably, many pathophysiological processes implicated in the development of AF and VF are linked to a mitochondrial dysfunction, which causes an altered calcium homeostasis, an excess of reactive oxygen species formation (oxidative stress), and alterations in the oxygen consumption. Mitochondria are considered to be a metabolic sink and [] the targets for suppressing arrhythmias [].
Despite the progress in the treatment of heart diseases and the management of arrhythmias, sudden cardiac death, occurring due to malignant ventricular arrhythmias, remains a major cause of mortality globally []. An implantable cardioverter defibrillator may be efficient in preventing sudden death due to VF when it occurs but cannot prevent VF development and/or its recurrence. This issue remains to be investigated to reduce the risk of an incident VF.
3.2. Thyroid Status Imbalance Promotes VF
In contrast to the prevalence of AF, the incidence of VF attributed solely to the hyperthyroid status is less common and registered with a frequency similar to that in the euthyroid population []. It is likely because VF is exceptional in those cases where TH levels are elevated but without the observation of an arrhythmogenic structural substrate or channelopathies []. Nevertheless, ventricular tachycardia has been registered in hyperthyroid patients suffering from Grave’s disease and it has been associated with the interaction of autoantibodies of the β1-adrenergic, the M2 muscarinic, and the TSH-receptors []. While thyroidectomy in Graves’ disease attenuates the occurrence of ventricular arrhythmias [], it seems likely that VF may occur in individuals with an altered thyroid status when accompanied by the presence of the autoantibodies.
Autoimmunity may alter the myocardial electrical properties promoting idiopathic ventricular arrhythmias [,]. Recent data strongly point out the implication of autoantibodies toward the β1 and β2 adrenergic or the M2 muscarinic receptors and the myosin heavy chain in the occurrence of cardiac rhythm disturbances [,,,,]. Anti-β adrenergic and anti-muscarinic receptor antibodies affect the myocardial electrophysiological properties and have been reported to be the independent predictors of sudden cardiac death in patients with various heart diseases []. The dysregulating effects of the autoantibodies against the calcium and potassium ion channels can play the basis for autoimmune phenocopies of genetic cardiac channelopathies []. Autoimmune cardiac channelopathies have been suggested as a novel mechanism in the development of cardiac arrhythmias [].
Occasionally, acute thyrotoxicosis accompanied by severe hypokalemia can induce a persistent ventricular tachycardia []. Unlike poor evidence in humans, the impact of TH on the development of VF in experimental animals is well documented [,,,] and associated with both the genomic and non-genomic TH actions []. Perhaps because of high TH dose, the oxidative stress-related impairment of ion and Cx43 channels is more pronounced in animal models.
Concerning TH, the prevalence of atrial versus ventricular arrhythmias may be explained by the chamber-related differences in the expression of ion and connexin channels, the duration of the effective refractory period, conduction velocity, and local activation time [,,], as well as in numerous signaling pathways [] and distinct tissue pro-fibrotic properties []. Certainly, hyperthyroidism promotes a myocardial electrical instability [] due to an increased excitability and shortening of the repolarization, thereby facilitating triggered activity and ventricular premature beats that often initiate malignant arrhythmias in a structurally altered heart []. TH may also affect ventricular arrhythmogenesis via an influence of the ANS [] as the density of the adrenergic binding sites has been shown to be enhanced by a chronic or an acute treatment with TH [,].
4. Thyroid Malignancies, Treatment, and Risk for Arrhythmias
4.1. Pathomechanisms, Incidence, and Treatment of Thyroid Carcinoma (TCA)
Thyroid dysfunction and diseases are more prevalent in women (about 5–20 times higher than those in men) and their incidence is more pronounced with age []. Likewise, the incidence of differentiated thyroid cancer (DTCA) increases in the population over 55 years []. Papillary and follicular thyroid carcinoma are the most common types of DTCA []. Undifferentiated medullary and anaplastic thyroid cancer have, however, a less favorable prognosis than that for DTCA ([].
Inflammation and oxidative stress are involved in the pathogenesis of cancer in a manner similar to that in cardiovascular diseases [,,,] by contributing to the initiation, progression, and complication of these diseases []. The genetic landscape is clearly evident and gene rearrangements are classified according to a spectrum of the RAS-like and BRAF-like tumors []. BRAFV600E mutation positivity and mutations in the telomerase reverse transcriptase promoter may also be prognostic in TCA progression or recurrence [,]. A mutational analysis helps to define the appropriate initial management, adjuvant therapy, surveillance protocols, and treatment [].
Standard management of the TCA patients includes a total thyroidectomy, a neck lymph node dissection (if indicated), and radioactive iodine (RAI) ablation followed by a TSH suppressive T4 therapy. Surgery is the primary crucial treatment to remove the tumor and the involved regional lymph nodes, to facilitate an accurate staging of the disease, to minimize the risk of disease recurrence and metastasis, and to facilitate a postoperative treatment with RAI and an accurate long-term surveillance for recurrence []. The accuracy of the post-radioiodine SPECT/CT predicts a long-term outcome of DTCA [,].
Total thyroidectomy followed by RAI has been associated with benefits in high-risk patients, as well as in the decrease in TCA recurrence. Current guidelines recommend an individualized approach for RAI indication [] because its benefit in low-risk patients was not supported by evidence [,,]. Despite the potential adverse effects of RAI, its application increases in patients with DTCA [,]. Decreased quality of life in TCA survivors has been recently reported [], but there is a lack of information about arrhythmias.
Post-surgery suppressive therapy with T4 downregulates the pituitary TSH that promotes growth of the residual malignant thyroid cells []. Thus, lowering the TSH levels with an exogenous T4 is involved in the long-term management of DTCA []. The benefit of this therapy has been established for high-risk but not for low-risk patients []. Long-term treatment with T4 affects the TH metabolism and results in a stable subclinical hyperthyroid state along with the down-regulation of deiodinases 1–2 and upregulation of deiodinase 3 []. Whether these patients exhibit cardiac rhythm disturbances is not known, while an inverse correlation between TSH levels and the risk of AF has been well established in a benign thyroid disease [].
Besides exogenous T4, TH analogs possessing a thyroidomimetic activity, such as triiodothyroacetic acid (an acetic acid metabolite of T3) and tetraiodothyroacetic acid (a derivate of T4), reduce the risk of cancer progression, enhance therapeutic effects, and suppress cancer recurrence []. However, all exogenous approaches exert adverse cardiovascular effects as indicated by earlier and recent studies [,,,,,]. It is challenging for cardiac arrhythmia screening.
The thyroid axis ‘set-points’ are significantly altered after a long-term T4 therapy and the dosage required for the TSH suppression may be reduced over time []. However, the appropriate degree of TSH suppression remains unsettled according to discrepancies among guidelines []. Nevertheless, TSH in the low-normal range <1 mIU/L seems to be preferable in most patients with DTCA []. In the absence of prospective trials, the discussion still remains concerning the extent of surgery, benefit and dosing of postoperative RAI, as well as the optimal level and duration of a TH-suppressive therapy []. The updated trials should also pay attention to arrhythmia risk stratification when considering predisposition to AF, including asymptomatic.
4.2. Thyroid Carcinoma Suppressive Therapy and Risk for Cardiac Arrhythmias
There is a link between AF and thyroid nodules, whereby cytokines and growth factors, such as IGF-1, EGF, and FGF, may be involved []. Moreover, TH regulate oxidative metabolism and plays an important role in the production of free oxygen radicals, thereby promoting oxidative stress. This event, as well as autoimmune thyroiditis, has been shown to be involved in the pathogenesis, as well as adverse effects in patients suffering from TCA [,,,]. Elderly persons, even clinically euthyroid but with low or low-normal TSH level, are at increased risk for AF [].
TCA is considered an independent predictor for AF along with well-known AF risk factors [,,]. Notably, the increased risk of AF in patients treated for DTCA but without pre-existing cardiovascular disease may warrant periodic screening for this arrhythmia []. This is in line with a recent study indicating that patients with DTCA along with TSH suppression below 0.1 mIU/L have a higher risk of AF []. Some trials did not find an association between the TSH level but have found an association between the cumulative dose of RAI and AF within the TCA cohort [].
According to a Swedish Nationwide Study, DTCA patients have a higher AF incidence compared to the general population, and females face a slightly higher incidence of cerebrovascular disease []. Thus, it appears relevant to pay attention to the AF risk factors as well as to an early identification of cardiac rhythm disturbances in TCA patients during their follow up (Figure 2). It may prevent AF-related complications and reduce the symptoms. The need for accurate risk stratification and long-term outcome data to support the treatment decisions is highly relevant for those with early-stage, very low-risk tumors, as well as for those with advanced and metastatic disease []. There is also limited information regarding the DTCA treatment-related morbidity associated with a long-term hypocalcemia [] that may provoke rhythm disturbances.

Figure 2.
Algorithm of the thyroid cancer therapy and possible risk for development of atrial fibrillation (AF) due to subclinical hyperthyroidism.
In the context of arrhythmias, an interesting question arises as to whether TCA patients during follow-up exhibit some specific risk factors for cardiac arrhythmias, such as the pro-arrhythmic β1-adrenergic and M2 muscarinic receptor autoantibodies or changes in the myocardial connexins. It has been shown that TSH and TH regulate the expression of connexins in a variety of target tissues, including the thyroid gland []. Reduced connexin-43 expression in the thyroid tissue differentiates TCA from a benign disease and provides clinical utility as a marker for malignancy [,], while decreased connexin-43 protein abundance in the cardiac tissue in the experimental hyperthyroid setting increases the propensity to AF and VF [,,,]. On the other hand, the upregulation of atrial connexin-40 precedes AF as well []. Alteration of both connexin-43 and connexin-40 abundance impairs an intercellular electrical coupling, thereby affecting the myocardial conduction and facilitating cardiac arrhythmias [,].
5. Cellular and Molecular Actions Potentially Involved in Pro-Arrhythmic Signaling of TH
TH are powerful modulators of electrical properties of the heart [,,,], thus playing an important role in the development of cardiac arrhythmias that occur due to electrical disturbances. Molecular targets for TH implicated in electrical instability that may facilitate AF or VF occurrence have been summarized in Table 1.

Table 1.
Cellular actions potentially involved in pro-arrhythmic signaling of TH in the heart.
5.1. Targeting Cardiac Ion Channels
Microarrays analyses showed that the hyperthyroid status increased the expression levels of genes encoding the voltage-gated potassium channel proteins, notably KCNA5 (Kv1.5) and KCNB1 (Kv2.1), that contributed to the main repolarizing K+ currents, as well as the HCN2 and HCN4 genes encoding pacemaker If channels []. In contrast, the expression of KCNQ1 (KvLQT1) and its regulator KCNE1 (minK) generating the slow component of cardiac delayed rectification, IKs current, was decreased. The up-regulation of Kv1.5 mRNA was greater in the atrium than that in the ventricle.
Electrophysiological studies revealed that the sinus tachycardia was related to the effect of TH on the rate of diastolic depolarization of the pacemaker cells via the increase in the pacemaker HCN2 (If) current [,]. In this context, it should be noted that the hyperpolarization-activated cyclic nucleotide-gated HCN channels isoforms are also expressed in the atrial and ventricular tissue; therefore, they may be implicated in promoting a triggered activity in a pathophysiological setting []. Indeed, TH can activate electrical triggers inducing an abnormal depolarization [] that often arises from the cardiomyocytes overlapping the pulmonary veins.
Enhanced triggered activity may increase the arrhythmogenic activity due to a higher incidence of DAD that often initiate AF []. Delayed-rectifier IKur and acetylcholine-regulated IKAch currents are present solely in the atrial tissue []. The shortening of the action potential duration (APD) in hyperthyroid rat atria was linked with a remarkably increased IKur and a decreased L-type Ca2+ current [,,], while the latter was increased in the ventricle []. Th shortening of the APD associated with decreased refractoriness has been considered as one of the main mechanisms for the risk of AF in hyperthyroidism [,,]. These conditions likely affect the P-wave duration and may increase the P-wave dispersion predisposing to an AF [].
It is noteworthy that the interaction of β1-adrenergic receptor autoantibodies with the target receptor amplifies the L-type Ca2+ current (ICa) and rapid delayed rectifier K+ currents (Iks, Ikr), which result in a decreased APD and the increase in the membrane potential during the plateau phase []. Interaction of the M2 muscarinic receptor autoantibodies with the target receptor inhibits ICa and increases the outward acetylcholine-regulated potassium current (IK,Ach), which results in a hyperpolarization and shortening of APD [].
5.2. Targeting Cardiac Ca2+ Handling
Thyroid status affects myocardial Ca2+ handling and intracellular free Ca2+ concentration by regulating the transcription of the genes involved in Ca2+ cycling []. Both gene expression and protein levels of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) increase in response to TH [], along with suppression of phospholamban expression []. It suggests an increase in Ca2+ uptake by sarcoplasmic reticulum whereby its Ca2+ overload may facilitate spontaneous Ca2+ leaks. An abnormal Ca2+ cycling and Ca2+ leak through the ryanodine receptor (RyR) in sarcoplasmic reticulum are involved in the occurrence of triggered activity such as EAD or DAD, followed by premature contraction []. DAD may occur during tachycardia in hyperthyroidism and contribute directly to increased susceptibility of the heart to AF and VF []. Moreover, TH increased the INa current, resulting in an increase in intracellular Ca2+ likely via an accelerated reverse mode of Na+/Ca2+ exchanger []. High diastolic Ca2+ and Ca2+ overloads inhibit intercellular communication mediated by connexin channels that may result in a slower or block of conduction promoting re-entrant arrhythmias [,].
5.3. Targeting Myocardial Metabolism, Structure, and Intercellular Coupling
TH induce a hyperdynamic cardiovascular state accompanied by an elevation of the left atrial pressure and increase in the size of the atria that facilitates a paroxysmal AF []. It suggests that, apart from the ion and Ca2+-handling alterations, AF occurrence is promoted by the presence of a structural arrhythmogenic substrate, such as hypertrophy. This is linked with alterations in the expression, phosphorylation, and distribution of the Cx40, Cx43, and Cx45 channels [,,,,,]. Impairment of the connexin channels deteriorates the electrical coupling required for an AP propagation, thereby affecting the myocardial conduction and promoting re-entry mechanisms []. Hyperthyroid rats exhibited a down-regulation of Cx43 in both the atrial and ventricular tissues and were prone to an electrically inducible AF and VF [,,]. In this context, it should be noted that functional phosphorylated forms of Cx43 were markedly suppressed in both the atria and ventricles of hyperthyroid rats. It can be attributed to the down-regulation of the protein kinase C epsilon (PKCε) that is suppressed by TH [,].
Moreover, a cardiac overload of various etiologies (including hyperthyroidism) is associated with an altered myocardial redox state and oxidative stress, which impairs the function of numerous target proteins including the ion and connexin channels, thereby facilitating the development of AF or VF [,].
5.4. Targeting ANS and RAAS
TH interact with the sympathetic nervous system by augmenting responsiveness to a sympathetic stimulation presumably via modulating the adrenergic receptor function and/or density. Myocardial adrenergic receptor binding sites have been shown to be enhanced by a chronic, as well as an acute treatment with TH []. There is also cross-talk between TH and the renin–angiotensin–aldosterone system []. TH activate RAAS through the TH response elements (TREs), thereby increasing the expression of mRNA for renin. Taken together, it seems realistic that ANS and RAAS may be additional players implicated in the development of arrhythmias in hyperthyroidism.
6. Conclusions
TH regulate multiple nuclear and extra-nuclear processes that operate to maintain the cardiac function. An overt or more frequent subclinical hyperthyroid state due to thyroid diseases disturb this regulation, promoting the development of cardiac arrhythmias, mostly AF. Disorders in the intracellular Ca2+ due to an altered Ca2+ handling, along with alteration of the expression and function of the HCN, Na+, K+, and Ca2+ channel and an impairment in the connexin channel-mediated cell-to-cell coupling, seem to be crucial factors in the proarrhythmic signaling of TH. Accordingly, it appears that the long-term subclinical hyperthyroid state including a TSH suppressive therapy with L-thyroxine may increase the risk for AF in post-thyroidectomy patients. Though relatively uncommon, perhaps because of a lack of real evidence due to limitations in the screening process [], awareness about potential interactions promoting AF, particularly asymptomatic, and to prevent undesirable consequences is required.
Author Contributions
Conceptualization, writing review, editing, funding acquisition, N.T.; Writing review, editing, funding acquisition, L.H.K.; Visualization, editing, P.H.; Editing, K.H.; Writing review, editing, funding acquisition, B.S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Slovak VEGA 2/0002/20, 2/0158/19, APVV 18-0548, EU ITMS 26230120009, and MEXT-Supported Program in Kagawa University.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Farré, J.; Wellens, H.J. Philippe Coumel: A founding father of modern arrhythmology. Europace 2004, 6, 464–465. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.F. Cardiac arrhythmias: What do we need to know about basic mechanisms? J. Cardiovasc. Electrophysiol. 1999, 10, 414–416. [Google Scholar] [PubMed]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.; Li, N.; Karck, M.; Wehrens, X.; Nattel, S.; Dobrev, D. Cellular and Molecular Mechanisms of Atrial Arrhythmogenesis in Patients with Paroxysmal Atrial Fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef]
- Tribulova, N.; Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Barancik, M. New aspects of pathogenesis of atrial fibrillation: Remodeling of intercalated discs. J. Physiol. Pharmacol. 2015, 66, 625–634. [Google Scholar]
- Tribulova, N.; Szeiffova Bacova, B.; Benova, T.; Viczenczova, C. Can we protect from malignant arrhythmias by modulation of cardiac cell-to-cell coupling? J. Electrocardiol. 2015, 48, 434–440. [Google Scholar] [CrossRef]
- Fabritz, L.; Schotten, U.; Kirchhof, P. ESC CardioMed, 3rd ed.; Camm, A.J., Lüsche, T.F., Maurer, G., Serruys, P.W., Eds.; Oxford University Press: Oxford, UK, 2018; ISBN 9780198784906. [Google Scholar]
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867. [Google Scholar] [CrossRef]
- Roney, C.H.; Wit, A.L.; Peters, N.S. Challenges Associated with Interpreting Mechanisms of AF. Arrhythmia Electrophysiol. Rev. 2019, 8, 273–284. [Google Scholar] [CrossRef]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef]
- Larsen, P. Thyroid-pituitary interaction: Feedback regulation of thyrotropin secretion by thyroid hormones. N. Engl. J. Med. 1982, 306, 23–32. [Google Scholar]
- Franklyn, J.A.; Boelaert, K. Thyrotoxicosis. Lancet 2012, 379, 1155–1166. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.J.; Yuan, X.; Li, C.; Sheng, H.; Su, B.; Sheng, C.; Qu, S.; Li, H. Hyperthyroidism caused by acquired immune deficiency syndrome. Eur. Rev. Med. Pharmacol. Sci. Hyperthyroid. 2014, 18, 875–879. [Google Scholar]
- Skelin, M.; Lucijanić, T.; Amidžić Klarić, D.; Rešić, A.; Bakula, M.; Liberati-Čizmek, A.M.; Gharib, H.; Rahelić, D. Factors Affecting Gastrointestinal Absorption of Levothyroxine: A Review. Clin. Ther. 2017, 39, 378–403. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Knezl, V.; Shainberg, A.; Seki, S.; Soukup, T. Thyroid hormones and cardiac arrhythmias. Vasc. Pharmacol. 2010, 52, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Collet, T.H.; Gussekloo, J.; Bauer, D.C.; Den Elzen, W.P.J.; Cappola, A.R.; Balmer, P.; Iervasi, G.; Åsvold, B.O.; Sgarbi, J.A.; Völzke, H.; et al. Subclinical hyperthyroidism and the risk of coronary heart disease and mortality. Arch. Intern. Med. 2012, 172, 799–809. [Google Scholar] [CrossRef]
- Cappola, A.R.; Desai, A.S.; Medici, M.; Cooper, L.S.; Egan, D.; Sopko, G.; Fishman, G.I.; Goldman, S.; Cooper, D.S.; Mora, S.; et al. Thyroid and Cardiovascular Disease: Research Agenda for Enhancing Knowledge, Prevention, and Treatment. Circulation 2019, 139, 2892–2909. [Google Scholar] [CrossRef]
- Hollowell, J.G.; Staehling, N.W.; Dana Flanders, W.; Harry Hannon, W.; Gunter, E.W.; Spencer, C.A.; Braverman, L.E. Serum TSH, T4, and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J. Clin. Endocrinol. Metab. 2002, 87, 489–499. [Google Scholar] [CrossRef]
- Portman, M.A. Thyroid hormone regulation of heart metabolism. Thyroid 2008, 18, 217–225. [Google Scholar] [CrossRef]
- Iordanidou, A.; Hadzopoulou-Cladaras, M.; Lazou, A. Non-genomic effects of thyroid hormone in adult cardiac myocytes: Relevance to gene expression and cell growth. Mol. Cell. Biochem. 2010, 340, 291–300. [Google Scholar] [CrossRef]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef]
- Jabbar, A.; Pingitore, A.; Pearce, S.H.S.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2016, 14, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Taha, W.; Kundumadam, S.; Khan, M. Atrial fibrillation and hyperthyroidism: A literature review. Indian Heart J. 2017, 69, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Ellervik, C.; Roselli, C.; Christophersen, I.E.; Alonso, A.; Pietzner, M.; Sitlani, C.M.; Trompet, S.; Arking, D.E.; Geelhoed, B.; Guo, X.; et al. Assessment of the relationship between genetic determinants of thyroid function and atrial fibrillation: A mendelian randomization study. JAMA Cardiol. 2019, 4, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, C.; Da Costa, B.R.; Collet, T.H.; Feller, M.; Floriani, C.; Bauer, D.C.; Cappola, A.R.; Heckbert, S.R.; Ceresini, G.; Gussekloo, J.; et al. Thyroid Function Within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation 2017, 136, 2100–2116. [Google Scholar] [CrossRef]
- Szeiffová Bačova, B.; Egan Beňová, T.; Viczenczová, C.; Soukup, T.; Rauchová, H.; Pavelka, S.; Knezl, V.; Barancík, M.; Tribulová, N. Cardiac connexin-43 and PKC signaling in rats with altered thyroid status without and with omega-3 fatty acids intake. Physiol. Res. 2016, 65, 77–90. [Google Scholar] [CrossRef]
- Bačová, B.S.; Vinczenzová, C.; Žurmanová, J.; Kašparová, D.; Knezl, V.; Beňová, T.E.; Pavelka, S.; Soukup, T.; Tribulová, N. Altered thyroid status affects myocardial expression of connexin-43 and susceptibility of rat heart to malignant arrhythmias that can be partially normalized by red palm oil intake. Histochem. Cell Biol. 2017, 147, 63–73. [Google Scholar] [CrossRef]
- Donzelli, R.; Colligiani, D.; Kusmic, C.; Sabatini, M.; Lorenzini, L.; Accorroni, A.; Nannipieri, M.; Saba, A.; Iervasi, G.; Zucchi, R. Effect of Hypothyroidism and Hyperthyroidism on Tissue Thyroid Hormone Concentrations in Rat. Eur. Thyroid J. 2016, 5, 27–34. [Google Scholar] [CrossRef][Green Version]
- Gil-Cayuela, C.; Roselló-LLetí, E.; Tarazón, E.; Ortega, A.; Sandoval, J.; Martínez-Dolz, L.; Cinca, J.; Jorge, E.; González-Juanatey, J.R.; Lago, F.; et al. Thyroid hormone biosynthesis machinery is altered in the ischemic myocardium: An epigenomic study. Int. J. Cardiol. 2017, 243, 27–33. [Google Scholar] [CrossRef]
- Heijman, J.; Voigt, N.; Ghezelbash, S.; Schirmer, I.; Dobrev, D. Calcium handling abnormalities as a target for Atrial fibrillation therapeutics: How close to clinical implementation? J. Cardiovasc. Pharmacol. 2015, 66, 515–522. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, W.; Thomson, J.K.; Gao, X.; DeMarco, D.M.; Carrillo, E.; Chen, B.; Wu, X.; Ginsburg, K.S.; Bakhos, M.; et al. Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ. Res. 2018, 122, 821–835. [Google Scholar] [CrossRef]
- Rahmutula, D.; Zhang, H.; Wilson, E.; Olgin, J. Absence of natriuretic peptide clearance receptor attenuates TGF-β1-induced selective atrial fibrosis and atrial fibrillation. Cardiovasc. Res. 2019, 115, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, F.; Schulte, J.S.; Gomes, B.; Zafeiriou, M.P.; Ratte, A.; Rathjens, F.; Fehrmann, E.; Scholz, B.; Voigt, N.; Müller, F.U.; et al. Atrial fibrillation and heart failure-associated remodeling of two-pore-domain potassium (K2P) channels in murine disease models: Focus on TASK-1. Basic Res. Cardiol. 2018, 113, 27. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Obata, K.; Ohmori, T.; Ishiwata, K.; Abe, M.; Hamaguchi, S.; Namekata, I.; Tanaka, H. Angiotensin II Induces Automatic Activity of the Isolated Guinea Pig Pulmonary Vein Myocardium through Activation of the IP₃ Receptor and the Na+-Ca2+ Exchanger. Int. J. Mol. Sci. 2019, 20, 1768. [Google Scholar] [CrossRef] [PubMed]
- Carballo, S.; Pfenniger, A.; Carballo, D.; Garin, N.; James, R.W.; Mach, F.; Shah, D.; Kwak, B.R. Differential association of Cx37 and Cx40 genetic variants in atrial fibrillation with and without underlying structural heart disease. Int. J. Mol. Sci. 2018, 19, 295. [Google Scholar] [CrossRef] [PubMed]
- Bai, D. Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 2014, 588, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Noureldin, M.; Chen, H.; Bai, D. Functional characterization of novel atrial fibrillation-linked GJA5 (CX40) mutants. Int. J. Mol. Sci. 2018, 19, 977. [Google Scholar] [CrossRef]
- Mann, I.; Sandler, B.; Linton, N.; Kanagaratnam, P. Drivers of atrial fibrillation: Theoretical considerations and practical concerns. Arrhythmia Electrophysiol. Rev. 2018, 7, 49–54. [Google Scholar] [CrossRef]
- Hohl, M.; Linz, B.; Böhm, M.; Linz, D. Obstructive sleep apnea and atrial arrhythmogenesis. Curr. Cardiol. Rev. 2014, 10, 362–368. [Google Scholar] [CrossRef]
- Wolke, C.; Bukowska, A.; Goette, A.; Lendeckel, U. Redox control of cardiac remodeling in atrial fibrillation. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1555–1565. [Google Scholar] [CrossRef]
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484. [Google Scholar] [CrossRef]
- Schmidt, C.; Wiedmann, F.; Voigt, N.; Zhou, X.B.; Heijman, J.; Lang, S.; Albert, V.; Kallenberger, S.; Ruhparwar, A.; Szabó, G.; et al. Upregulation of K2P 3.1 K + Current Causes Action Potential Shortening in Patients with Chronic Atrial Fibrillation. Circulation 2015, 132, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Seyed Ahmadi, S.; Svensson, A.M.; Pivodic, A.; Rosengren, A.; Lind, M. Risk of atrial fibrillation in persons with type 2 diabetes and the excess risk in relation to glycaemic control and renal function: A Swedish cohort study. Cardiovasc. Diabetol. 2020, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lin, Y. The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome. Int. J. Mol. Sci. 2020, 21, 891. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Choi, E.; Cha, M.J.; Hwang, K.C. Looking into a conceptual framework of ROS–miRNA–Atrial fibrillation. Int. J. Mol. Sci. 2014, 15, 21754–21776. [Google Scholar] [CrossRef]
- Liew, R.; Khairunnisa, K.; Gu, Y.; Tee, N.; Yin, N.O.; Naylynn, T.M.; Moe, K.T. Role of tumor necrosis factor-α in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ. J. 2013, 77, 1171–1179. [Google Scholar] [CrossRef]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in Atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Lebesgue, N.; Beavers, D.L.; Alsina, K.M.; Damen, J.M.A.; Voigt, N.; Dobrev, D.; Wehrens, X.H.T.; Scholten, A. Alterations in the interactome of serine/threonine protein phosphatase type-1 in atrial fibrillation patients. J. Am. Coll. Cardiol. 2015, 65, 163–173. [Google Scholar] [CrossRef][Green Version]
- Ghezelbash, S.; Molina, C.E.; Dobrev, D. Altered atrial metabolism: An underappreciated contributor to the initiation and progression of atrial fibrillation. J. Am. Heart Assoc. 2015, 4, e001808. [Google Scholar] [CrossRef]
- Hu, B.; Sun, Y.; Li, S.; Sun, J.; Liu, T.; Wu, Z.; Feng, L. Association of β1-Adrenergic, M2-Muscarinic Receptor Autoantibody with Occurrence and Development of Nonvalvular Atrial Fibrillation. Pace Pacing Clin. Electrophysiol. 2016, 39, 1379–1387. [Google Scholar] [CrossRef]
- Capecchi, P.L.; Laghi-Pasini, F.; El-Sherif, N.; Qu, Y.; Boutjdir, M.; Lazzerini, P.E. Autoimmune and inflammatory K+ channelopathies in cardiac arrhythmias: Clinical evidence and molecular mechanisms. Heart Rhythm 2019, 16, 1273–1280. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F.; Boutjdir, M. Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat. Rev. Cardiol. 2017, 14, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Chera, H.; Nagar, M.; Richler, A.; Pourriahi, M.; Al-Sadawi, M.; Gunsburg, M.; Shoenfeld, Y.; Rosen, Y. Autoantibodies for Cardiac Channels and Sudden Cardiac Death and its Relationship to Autoimmune Disorders. Curr. Cardiol. Rev. 2018, 15, 49–54. [Google Scholar] [CrossRef]
- Qu, Y.S.; Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F.; El Sherif, N.; Boutjdir, M. Autoimmune Calcium Channelopathies and Cardiac Electrical Abnormalities. Front. Cardiovasc. Med. 2019, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Effect of catheter ablation on pre-existing abnormalities of left atrial systolic, diastolic, and neurohormonal functions in patients with chronic heart failure and atrial fibrillation. Eur. Heart J. 2019, 40, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.; Perera, T.; Elliott, A.D.; Twomey, D.J.; Kumar, S.; Munwar, D.A.; Khokhar, K.B.; Thiyagarajah, A.; Middeldorp, M.E.; Nalliah, C.J.; et al. Subclinical device-detected atrial fibrillation and stroke risk: A systematic review and meta-analysis. Eur. Heart J. 2018, 39, 1407–1415. [Google Scholar] [CrossRef]
- Brandes, A.; Smit, M.D.; Nguyen, B.O.; Rienstra, M.; Van Gelder, I.C. Risk factor management in atrial fibrillation. Arrhythmia Electrophysiol. Rev. 2018, 7, 118–127. [Google Scholar] [CrossRef]
- Weil, B.; Ozcan, C. Cardiomyocyte Remodeling in Atrial Fibrillation and Hibernating Myocardium: Shared Pathophysiologic Traits Identify Novel Treatment Strategies? BioMed Res. Int. 2015, 2015, 587361. [Google Scholar] [CrossRef]
- Ali, R.L.; Hakim, J.B.; Boyle, P.M.; Zahid, S.; Sivasambu, B.; Marine, J.E.; Calkins, H.; Trayanova, N.A.; Spragg, D.D. Arrhythmogenic propensity of the fibrotic substrate after atrial fibrillation ablation: A longitudinal study using magnetic resonance imaging-based atrial models. Cardiovasc. Res. 2019, 115, 1757–1765. [Google Scholar] [CrossRef]
- Sohinki, D.; Stavrakis, S. New approaches for treating atrial fibrillation: Focus on autonomic modulation. Trends Cardiovasc. Med. 2019. [Google Scholar] [CrossRef]
- Odening, K.E.; Deiß, S.; Dilling-Boer, D.; Didenko, M.; Eriksson, U.; Nedios, S.; Ng, F.S.; Roca Luque, I.; Sanchez Borque, P.; Vernooy, K.; et al. Mechanisms of sex differences in atrial fibrillation: Role of hormones and differences in electrophysiology, structure, function, and remodelling. Europace 2019, 21, 366–376. [Google Scholar] [CrossRef]
- Diederichsen, S.Z.; Haugan, K.J.; Brandes, A.; Lanng, M.B.; Graff, C.; Krieger, D.; Kronborg, C.; Holst, A.G.; Køber, L.; Højberg, S.; et al. Natural History of Subclinical Atrial Fibrillation Detected by Implanted Loop Recorders. J. Am. Coll. Cardiol. 2019, 74, 2771–2781. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, W.F.; Yong, J.H.E.; Sandhu, R.K.; Gladstone, D.J.; Simek, K.; Liu, Y.Y.; Quinn, F.R.; Tytus, R.; Zizzo, D.; Henein, S.; et al. Prevalence of undiagnosed atrial fibrillation in elderly individuals and potential cost-effectiveness of non-invasive ambulatory electrocardiographic screening: The ASSERT-III study. J. Electrocardiol. 2020, 58, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Matusik, P.T. Biomarkers and cardiovascular risk stratification. Eur. Heart J. 2019, 40, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, J.; Andersson, L.; Mirskaya, M.; Rosenqvist, M. Stepwise screening of atrial fibrillation in a 75-year-old population: Implications for stroke prevention. Circulation 2013, 127, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Chua, W.; Purmah, Y.; Cardoso, V.R.; Gkoutos, G.V.; Tull, S.P.; Neculau, G.; Thomas, M.R.; Kotecha, D.; Lip, G.Y.H.; Kirchhof, P.; et al. Data-driven discovery and validation of circulating blood-based biomarkers associated with prevalent atrial fibrillation. Eur. Heart J. 2019, 40, 1268–1276. [Google Scholar] [CrossRef]
- Patton, K.K.; Heckbert, S.R.; Alonso, A.; Bahrami, H.; Lima, J.A.C.; Burke, G.; Kronmal, R.A. N-terminal pro-B-type natriuretic peptide as a predictor of incident atrial fibrillation in the Multi-Ethnic study of atherosclerosis: The effects of age, sex and ethnicity. Heart 2013, 99, 1832–1836. [Google Scholar] [CrossRef]
- Kemp Gudmundsdottir, K.; Fredriksson, T.; Svennberg, E.; Al-Khalili, F.; Friberg, L.; Frykman, V.; Hijazi, Z.; Rosenqvist, M.; Engdahl, J. Stepwise mass screening for atrial fibrillation using N-terminal B-type natriuretic peptide: The STROKESTOP II study. Europace 2020, 22, 24–32. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Mikhailidis, D.P.; Rysz, J.; Banach, M. The mechanisms of atrial fibrillation in hyperthyroidism. Thyroid Res. 2009, 2, 4. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, S.A.; Chen, Y.J.; Chang, M.S.; Chan, P.; Lin, C.I. Effects of thyroid hormone on the arrhythmogenic activity of pulmonary vein cardiomyocytes. J. Am. Coll. Cardiol. 2002, 39, 366–372. [Google Scholar] [CrossRef]
- Yu, Z.; Huang, C.X.; Wang, S.Y.; Wang, T.; Xu, L. Thyroid hormone predisposes rabbits to atrial arrhythmias by shortening monophasic action period and effective refractory period: Results from an in vivo study. J. Endocrinol. Investig. 2009, 32, 253–257. [Google Scholar] [CrossRef]
- Gen, R.; Akbay, E.; Çamsari, A.; Özcan, T. P-wave dispersion in endogenous and exogenous subclinical hyperthyroidism. J. Endocrinol. Investig. 2010, 33, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Vadiveloo, T.; Donnan, P.T.; Cochrane, L.; Leese, G.P. The Thyroid Epidemiology, Audit, and Research Study (TEARS): Morbidity in patients with endogenous subclinical hyperthyroidism. J. Clin. Endocrinol. Metab. 2011, 96, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Chaker, L.; Heeringa, J.; Dehghan, A.; Medici, M.; Visser, W.E.; Baumgartner, C.; Hofman, A.; Rodondi, N.; Peeters, R.P.; Franco, O.H. Normal thyroid function and the risk of atrial fibrillation: The Rotterdam study. J. Clin. Endocrinol. Metab. 2015, 100, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Turan, E.; Can, I.; Turan, Y.; Uyar, M.; Cakır, M. Comparison of cardiac arrhythmia types between hyperthyroid patients with graves’ disease and toxic nodular goiter. Acta Endocrinol. (Buchar.) 2018, 14, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, H.; Huang, J.; Luo, D.; Tang, Y.; Yu, R.; Huang, H. Case report of recurrent atrial fibrillation induced by thyrotropin-secreting pituitary adenoma with Graves’ disease. Med. (Baltim.) 2018, 97, e11047. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.M.; Mohamed, H.E.; Noureldine, S.I.; Nazari-Shafti, T.Z.; Thethi, T.K.; Kandil, E. Impact of thyroidectomy on cardiac manifestations of Graves’ disease. Laryngoscope 2016, 126, 1256–1259. [Google Scholar] [CrossRef]
- Weetman, A.P. Graves’ disease following immune reconstitution or immunomodulatory treatment: Should we manage it any differently? Clin. Endocrinol. 2014, 80, 629–632. [Google Scholar] [CrossRef]
- Stavrakis, S.; Yu, X.; Patterson, E.; Huang, S.; Hamlett, S.R.; Chalmers, L.; Pappy, R.; Cunningham, M.W.; Morshed, S.A.; Davies, T.F.; et al. Activating Autoantibodies to the Beta-1 Adrenergic and M2 Muscarinic Receptors Facilitate Atrial Fibrillation in Patients With Graves’ Hyperthyroidism. J. Am. Coll. Cardiol. 2009, 54, 1309–1316. [Google Scholar] [CrossRef]
- Nussinovitch, U.; Shoenfeld, Y. The Diagnostic and clinical significance of anti-muscarinic receptor autoantibodies. Clin. Rev. Allergy Immunol. 2012, 42, 298–308. [Google Scholar] [CrossRef]
- Galloway, A.; Li, H.; Vanderlinde-Wood, M.; Khan, M.; Benbrook, A.; Liles, C.; Zillner, C.; Rao, V.; Cunningham, M.W.; Yu, X.; et al. Activating autoantibodies to the β1/2-adrenergic and M2 muscarinic receptors associate with atrial tachyarrhythmias in patients with hyperthyroidism. Endocrine 2015, 49, 457–463. [Google Scholar] [CrossRef]
- Li, H.; Murphy, T.; Zhang, L.; Huang, B.; Veitla, V.; Scherlag, B.J.; Kem, D.C.; Yu, X. β1-Adrenergic and M2 muscarinic autoantibodies and thyroid hormone facilitate induction of atrial fibrillation in male rabbits. Endocrinology 2016, 157, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Becker, N.P.; Müller, J.; Göttel, P.; Wallukat, G.; Schimke, I. Cardiomyopathy—An approach to the autoimmune background. Autoimmun. Rev. 2017, 16, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; Shoenfeld, Y. The clinical significance of Anti-Beta-1 adrenergic receptor autoantibodies in cardiac disease. Clin. Rev. Allergy Immunol. 2013, 44, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, M.U.; Gurses, K.M.; Kocyigit, D.; Kesikli, S.A.; Dural, M.; Evranos, B.; Yorgun, H.; Sahiner, L.; Kaya, E.B.; Oto, M.A.; et al. Cardiac autoantibody levels predict recurrence following cryoballoon-based pulmonary vein isolation in paroxysmal atrial fibrillation patients. J. Cardiovasc. Electrophysiol. 2015, 26, 615–621. [Google Scholar] [CrossRef]
- Pfenniger, A.; Arora, R. Beyond beta-blockers: Targeting the sympathetic nervous system for the prevention and treatment of atrial fibrillation. Cardiovasc. Res. 2019, 115, 1940–1942. [Google Scholar] [CrossRef]
- Bacova, B.S.; Radosinska, J.; Wallukat, G.; Barancik, M.; Wallukat, A.; Knezl, V.; Sykora, M.; Paulis, L.; Tribulova, N. Suppression of β1-adrenoceptor autoantibodies is involved in the antiarrhythmic effects of omega-3 fatty acids in male and female hypertensive rats. Int. J. Mol. Sci. 2020, 21, 526. [Google Scholar] [CrossRef]
- Tang, R.B.; Liu, D.L.; Dong, J.Z.; Liu, X.P.; Long, D.Y.; Yu, R.H.; Hu, F.L.; Wu, J.H.; Liu, X.H.; Ma, C.S. High-normal thyroid function and risk of recurrence of atrial fibrillation after catheter ablation. Circ. J. 2010, 74, 1317–1321. [Google Scholar] [CrossRef]
- Sousa, P.A.; Providência, R.; Albenque, J.P.; Khoueiry, Z.; Combes, N.; Combes, S.; Boveda, S. Impact of Free Thyroxine on the Outcomes of Left Atrial Ablation Procedures. Am. J. Cardiol. 2015, 116, 1863–1868. [Google Scholar] [CrossRef]
- Morishima, I.; Okumura, K.; Morita, Y.; Kanzaki, Y.; Takagi, K.; Yoshida, R.; Nagai, H.; Ikai, Y.; Furui, K.; Yoshioka, N.; et al. High-normal thyroid-stimulating hormone shows a potential causal association with arrhythmia recurrence after catheter ablation of atrial fibrillation. J. Am. Heart Assoc. 2018, 7, e009158. [Google Scholar] [CrossRef]
- Wei, S.B.; Wang, W.; Liu, N.; Chen, J.; Guo, X.Y.; Tang, R.B.; Yu, R.H.; Long, D.Y.; Sang, C.H.; Jiang, C.X.; et al. U-shaped association between serum free triiodothyronine and recurrence of atrial fibrillation after catheter ablation. J. Interv. Card. Electrophysiol. 2018, 51, 263–270. [Google Scholar] [CrossRef]
- Kim, K.H.; Mohanty, S.; Mohanty, P.; Trivedi, C.; Morris, E.H.; Santangeli, P.; Bai, R.; Al-Ahmad, A.; Burkhardt, J.D.; Gallinghouse, J.G.; et al. Prevalence of right atrial non-pulmonary vein triggers in atrial fibrillation patients treated with thyroid hormone replacement therapy. J. Interv. Card. Electrophysiol. 2017, 49, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P. Antiarrhythmic therapy in 2014: Contemporary approaches to treating arrhythmias. Nat. Rev. Cardiol. 2015, 12, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Bacharova, L. Missing link between molecular aspects of ventricular arrhythmias and QRS complex morphology in left ventricular hypertrophy. Int. J. Mol. Sci. 2019, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Huang, H.X.; Liu, P.; Du, Y.H.; Wang, P.; Wang, W.; Wu, Y.; Wang, L.; Ma, C.S.; Liu, H.R. β1-Adrenoceptor autoantibodies increase the susceptibility to ventricular arrhythmias involving abnormal repolarization in guinea-pigs. Exp. Physiol. 2017, 102, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, J.L.; Graff, C.; Ellervik, C.; Kanters, J.K. Electrocardiographic effect of thyroid hormones. J. Electrocardiol. 2019, 57, S108–S109. [Google Scholar] [CrossRef]
- Akar, F.G.; O’Rourke, B. Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol. Ther. 2011, 131, 287–294. [Google Scholar] [CrossRef]
- Akar, F.G. Mitochondrial targets for arrhythmia suppression: Is there a role for pharmacological intervention? J. Interv. Card. Electrophysiol. 2013, 37, 249–258. [Google Scholar] [CrossRef]
- Nadkarni, P.J.; Sharma, M.; Zinsmeister, B.; Wartofsky, L.; Burman, K.D. Thyrotoxicosis-induced ventricular arrhythmias. Thyroid 2008, 18, 1111–1114. [Google Scholar] [CrossRef]
- Eisen, A.; Arnson, Y.; Dovrish, Z.; Hadary, R.; Amital, H. Arrhythmias and Conduction Defects in Rheumatological Diseases-A Comprehensive Review. Semin. Arthritis Rheum. 2009, 39, 145–156. [Google Scholar] [CrossRef]
- Lee, H.C.; Huang, K.T.L.; Wang, X.L.; Shen, W.K. Autoantibodies and cardiac arrhythmias. Heart Rhythm 2011, 8, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Brisinda, D.; Sorbo, A.R.; Venuti, A.; Ruggieri, M.P.; Manna, R.; Fenici, P.; Wallukat, G.; Hoebeke, J.; Frustaci, A.; Fenici, R. Anti-β-adrenoceptors autoimmunity causing “idiopathic” arrhythmias and cardiomyopathy. Circ. J. 2012, 76, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Ryabkova, V.A.; Shubik, Y.V.; Erman, M.V.; Churilov, L.P.; Kanduc, D.; Shoenfeld, Y. Lethal immunoglobulins: Autoantibodies and sudden cardiac death. Autoimmun. Rev. 2019, 18, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.H.; Su, Y.J. Thyrotoxic periodic paralysis with ventricular tachycardia. J. Electrocardiol. 2019, 54, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.J.; Dai, D.Z.; Dai, Y. Up-regulated inflammatory factors endothelin, NFκB, TNFα and iNOS involved in exaggerated cardiac arrhythmias in l-thyroxine-induced cardiomyopathy are suppressed by darusentan in rats. Life Sci. 2006, 79, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Mitasikova, M.; Dlugosova, K.; Okruhlicova, L.; Imanaga, I.; Ogawa, K.; Weismann, P.; Tribulova, N. Thyroid hormones suppress ε-PKC signalling, down-regulate connexin-43 and increase lethal arrhythmia susceptibility in non-diabetic and diabetic rat hearts. J. Physiol. Pharmacol. 2008, 59, 271–285. [Google Scholar]
- Bačová, B.; Viczenczová, C.; Žurmanová, J.; Kašparová, D.; Knezl, V.; Beňová, T.; Pavelka, S.; Soukup, T.; Tribulová, N. Susceptibility of rats with altered thyroid status to malignant arrhythmias is related mainly to myocardial levels of connexin-43 and can be partially ameliorated by supplementation with red palm oil. Exp. Clin. Cardiol. 2013, 18, 41–46. [Google Scholar]
- Knezl, V.; Soukup, T.; Okruhlicová, L.; Slezák, J.; Tribulová, N. Thyroid hormones modulate occurrence and termination of ventricular fibrillation by both long-term and acute actions. Physiol. Res. 2008, 57, 91–96. [Google Scholar]
- Caves, R.E.; Cheng, H.; Choisy, S.C.; Gadeberg, H.C.; Bryant, S.M.; Hancox, J.C.; James, A.F. Atrial-ventricular differences in rabbit cardiac voltage-gated Na+ currents: Basis for atrial-selective block by ranolazine. Heart Rhythm 2017, 14, 1657–1664. [Google Scholar] [CrossRef]
- Lane, J.D.; Montaigne, D.; Tinker, A. Tissue-Level Cardiac Electrophysiology Studied in Murine Myocardium Using a Microelectrode Array: Autonomic and Thermal Modulation. J. Membr. Biol. 2017, 250, 471–481. [Google Scholar] [CrossRef]
- Johnson, E.K.; Matkovich, S.J.; Nerbonne, J.M. Regional Differences in mRNA and lncRNA Expression Profiles in Non-Failing Human Atria and Ventricles. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; De Martino Rosaroll, P.; Piro, M.C.; De Leo, T. Electrophysiological properties of the hyperthyroid rat heart. Arch. Int. Physiol. Biochim. Biophys. 1994, 102, 153–159. [Google Scholar]
- Buscemi, S.; Verga, S.; Cottone, S.; Andronico, G.; D’Orio, L.; Mannino, V.; Panzavecchia, D.; Vitale, F.C.G. Favorable clinical heart and bone effects of anti-thyroid drug therapy in endogenous subclinical hyperthyroidism. J. Endocrinol. Investig. 2007, 30, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Polikar, R.; Burger, A.; Scherrer, U.; Nicod, P. The Thyroid and the Heart. Circulation 1993, 87, 1435–1441. [Google Scholar] [CrossRef]
- Gross, G.; Lues, I. Thyroid-dependent alterations of myocardial adrenoceptors and adrenoceptor-mediated responses in the rat. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1985, 329, 427–439. [Google Scholar] [CrossRef]
- Tielens, E.T.; Forder, J.R.; Chatham, J.C.; Marrelli, S.P.; Ladenson, P.W. Acute L-triiodothyronine administration potentiates inotropic responses to β-adrenergic stimulation in the isolated perfused rat heart. Cardiovasc. Res. 1996, 32, 306–310. [Google Scholar] [CrossRef][Green Version]
- Gietka-Czernel, M. The thyroid gland in postmenopausal women: Physiology and diseases. Prz. Menopauzalny 2017, 16, 33–37. [Google Scholar] [CrossRef]
- Wang, T.S.; Sosa, J.A. Thyroid surgery for differentiated thyroid cancer—Recent advances and future directions. Nat. Rev. Endocrinol. 2018, 14, 670–683. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef]
- Shin, H.J.; Hwang, K.A.; Choi, K.C. Antitumor effect of various phytochemicals on diverse types of thyroid cancers. Nutrients 2019, 11, 125. [Google Scholar] [CrossRef]
- Wang, D.; Feng, J.F.; Zeng, P.; Yang, Y.H.; Luo, J.; Yang, Y.W. Total oxidant/antioxidant status in sera of patients with thyroid cancers. Endocr. Relat. Cancer 2011, 18, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Hosseini-Zijoud, S.M.; Ebadi, S.A.; Goodarzi, M.T.; Hedayati, M.; Abbasalipourkabir, R.; Mahjoob, M.P.; Poorolajal, J.; Zicker, F.; Sheikh, N. Lipid peroxidation and antioxidant status in patients with medullary thyroid carcinoma: A case-control study. J. Clin. Diagn. Res. 2016, 10, BC04–BC07. [Google Scholar] [PubMed]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and methods to measure oxidative stress in clinical samples: Research applications in the cancer field. Oxidative Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed]
- Macejova, D.; Podoba, J.; Toporova, L.; Grigerova, M.; Kajo, K.; Machalekova, K.; Brtko, J. Causal associations of autoimmune thyroiditis and papillary thyroid carcinoma: mRNA expression of selected nuclear receptors and other molecular targets. Oncol. Lett. 2019, 18, 4270–4277. [Google Scholar] [CrossRef]
- Libby, P.; Kobold, S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases—Expanding the concept of cardio-oncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef]
- Kebebew, E. Thyroid Cancer: Is It All in the Genes? J. Natl. Cancer Inst. 2018, 110, 327–328. [Google Scholar] [CrossRef]
- Takacsova, E.; Kralik, R.; Waczulikova, I.; Zavodna, K.; Kausitz, J. A different prognostic value of BRAFV600E mutation positivity in various age groups of patients with papillary thyroid cancer. Neoplasma 2017, 64, 156–164. [Google Scholar] [CrossRef]
- Donati, B.; Ciarrocchi, A. Telomerase and telomeres biology in thyroid cancer. Int. J. Mol. Sci. 2019, 20, 2887. [Google Scholar] [CrossRef]
- McIver, B.; Freeman, J.; Shah, J.P.; Shaha, A.R.; Haugen, B.; Cohen, E.; Witterick, I.J.; Randolph, G.W. Summary of the third world congress on thyroid cancer. Thyroid 2018, 28, 1401–1405. [Google Scholar] [CrossRef]
- Callender, G.G.; Carling, T.; Christison-Lagay, E.; Udelsman, R. Surgery for thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2014, 43, 443–458. [Google Scholar] [CrossRef]
- Szujo, S.; Sira, L.; Bajnok, L.; Bodis, B.; Gyory, F.; Nemes, O.; Rucz, K.; Kenyeres, P.; Valkusz, Z.; Sepp, K.; et al. The impact of post-radioiodine therapy SPECT/CT on early risk stratification in differentiated thyroid cancer; a bi-institutional study. Oncotarget 2017, 8, 79825–79834. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Carhill, A.A.; Litofsky, D.R.; Ross, D.S.; Jonklaas, J.; Cooper, D.S.; Brierley, J.D.; Ladenson, P.W.; Ain, K.B.; Fein, H.G.; Haugen, B.R.; et al. Long-term outcomes following therapy in differentiated thyroid carcinoma: NTCTCS registry analysis 1987–2012. J. Clin. Endocrinol. Metab. 2015, 100, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Ballal, S.; Soundararajan, R.; Garg, A.; Chopra, S.; Bal, C. Intermediate-risk differentiated thyroid carcinoma patients who were surgically ablated do not need adjuvant radioiodine therapy: Long-term outcome study. Clin. Endocrinol. 2016, 84, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, P.; Guarda, F.J.; Jaimovich, R.; Droppelmann, N.; González, H.; Domínguez, J.M. Radioactive Iodine Administration Is Associated with Persistent Related Symptoms in Patients with Differentiated Thyroid Cancer. Int. J. Endocrinol. 2016, 2016, 2586512. [Google Scholar] [CrossRef] [PubMed]
- Haymart, M.R.; Banerjee, M.; Stewart, A.K.; Koenig, R.J.; Birkmeyer, J.D.; Griggs, J.J. Use of radioactive iodine for thyroid cancer. JAMA J. Am. Med. Assoc. 2011, 306, 721–728. [Google Scholar] [CrossRef]
- Lee, Y.S.; Chang, H.S.; Park, C.S. Changing trends in the management of well-differentiated thyroid carcinoma in Korea. Endocr. J. 2016, 63, 515–521. [Google Scholar] [CrossRef]
- Aschebrook-Kilfoy, B.; James, B.; Nagar, S.; Kaplan, S.; Seng, V.; Ahsan, H.; Angelos, P.; Kaplan, E.L.; Guerrero, M.A.; Kuo, J.H.; et al. Risk Factors for Decreased Quality of Life in Thyroid Cancer Survivors: Initial Findings from the North American Thyroid Cancer Survivorship Study. Thyroid 2015, 25, 1313–1321. [Google Scholar] [CrossRef]
- Hesselink, E.N.K.; Lefrandt, J.D.; Schuurmans, E.P.; Burgerhof, J.G.M.; Groen, B.; Gansevoort, R.T.; Van Der Horst-Schrivers, A.N.A.; Dullaart, R.P.F.; Van Gelder, I.C.; Brouwers, A.H.; et al. Increased risk of atrial fibrillation after treatment for differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, 4563–4569. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Molecular functions of thyroid hormone signaling in regulation of cancer progression and anti-apoptosis. Int. J. Mol. Sci. 2019, 20, 4986. [Google Scholar] [CrossRef]
- Verburg, F.A.; Smit, J.W.A.; Grelle, I.; Visser, T.J.; Peeters, R.P.; Reiners, C. Changes within the thyroid axis after long-term TSH-suppressive levothyroxine therapy. Clin. Endocrinol. 2012, 76, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Selmer, C.; Olesen, J.B.; Hansen, M.L.; Lindhardsen, J.; Olsen, A.M.S.; Madsen, J.C.; Faber, J.; Hansen, P.R.; Pedersen, O.D.; Torp-Pedersen, C.; et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: A large population cohort study. BMJ (Online) 2012, 345, e7895. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Sudha, T.; Lin, H.Y.; Mousa, S.A. Thyroid hormone, hormone analogs, and angiogenesis. Compr. Physiol. 2016, 6, 353–362. [Google Scholar]
- Horne, M.K.; Singh, K.K.; Rosenfeld, K.G.; Wesley, R.; Skarulis, M.C.; Merryman, P.K.; Cullinane, A.; Costello, R.; Patterson, A.; Eggerman, T.; et al. Is thyroid hormone suppression therapy prothrombotic? J. Clin. Endocrinol. Metab. 2004, 89, 4469–4473. [Google Scholar] [CrossRef]
- Miyakawa, M. Effects of thyrotropin-suppressive therapy in patients with well-differentiated thyroid carcinoma. Nihon Rinsho Jpn. J. Clin. Med. 2007, 65, 2073–2077. [Google Scholar]
- Biondi, B.; Cooper, D.S. Benefits of thyrotropin suppression versus the risks of adverse effects in differentiated thyroid cancer. Thyroid 2010, 20, 135–146. [Google Scholar] [CrossRef]
- Klein Hesselink, E.N.; Klein Hesselink, M.S.; De Bock, G.H.; Gansevoort, R.T.; Bakker, S.J.L.; Vredeveld, E.J.; Van Der Horst-Schrivers, A.N.A.; Van Der Horst, I.C.C.; Kamphuisen, P.W.; Plukker, J.T.M.; et al. Long-term cardiovascular mortality in patients with differentiated thyroid carcinoma: An observational study. J. Clin. Oncol. 2013, 31, 4046–4053. [Google Scholar] [CrossRef]
- Abdulrahman, R.M.; Delgado, V.; Hoftijzer, H.C.; Ng, A.C.T.; Ewe, S.H.; Marsan, N.A.; Holman, E.R.; Hovens, G.C.; Corssmit, E.P.; Romijn, J.A.; et al. Both exogenous subclinical hyperthyroidism and short-term overt hypothyroidism affect myocardial strain in patients with differentiated thyroid carcinoma. Thyroid 2011, 21, 471–476. [Google Scholar] [CrossRef]
- Verburg, F.A.; Mäder, U.; Grelle, I.; Visser, T.J.; Peeters, R.P.; Smit, J.W.A.; Reiners, C. The thyroid axis “setpoints” are significantly altered after long-term suppressive LT4 therapy. Horm. Metab. Res. 2014, 46, 794–799. [Google Scholar] [CrossRef][Green Version]
- Pajamäki, N.; Metso, S.; Hakala, T.; Ebeling, T.; Huhtala, H.; Ryödi, E.; Sand, J.; Jukkola-Vuorinen, A.; Kellokumpu-Lehtinen, P.L.; Jaatinen, P. Long-term cardiovascular morbidity and mortality in patients treated for differentiated thyroid cancer. Clin. Endocrinol. 2018, 88, 303–310. [Google Scholar] [CrossRef]
- Bindi, M.; Moruzzo, D.; Rosada, J.; Castiglioni, M.; Romanelli, A.M. Atrial fibrillation and thyroid nodules. Recenti Progress. Med. 2011, 102, 17–19. [Google Scholar]
- Sawin, C.T.; Geller, A.; Wolf, P.A.; Belanger, A.J.; Baker, E.; Bacharach, P.; Wilson, P.W.; Benjamin, E.J. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N. Engl. J. Med. 1994, 331, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Abonowara, A.; Quraishi, A.; Sapp, J.L.; Alqambar, M.H.; Saric, A.; O’Connell, C.M.; Rajaraman, M.M.; Hart, R.D.; Imran, S.A. Prevalence of atrial fibrillation in patients taking TSH suppression therapy for management of thyroid cancer. Clin. Investig. Med. 2012, 35, 152–156. [Google Scholar] [CrossRef]
- Xia, Q.; Dong, S.; Da Bian, P.; Wang, J.; Li, C.J. Effects of endocrine therapy on the prognosis of elderly patients after surgery for papillary thyroid carcinoma. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Toulis, K.A.; Viola, D.; Gkoutos, G.; Keerthy, D.; Boelaert, K.; Nirantharakumar, K. Risk of incident circulatory disease in patients treated for differentiated thyroid carcinoma with no history of cardiovascular disease. Clin. Endocrinol. 2019, 91, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Zoltek, M.; Andersson, T.M.L.; Hedman, C.; Ihre-Lundgren, C.; Nordenvall, C. Cardiovascular Incidence in 6900 Patients with Differentiated Thyroid Cancer: A Swedish Nationwide Study. World J. Surg. 2020, 44, 436–441. [Google Scholar] [CrossRef]
- Parker, W.A.; Edafe, O.; Balasubramanian, S.P. Long-term treatment-related morbidity in differentiated thyroid cancer: A systematic review of the literature. Pragmatic Obs. Res. 2017, 8, 57–67. [Google Scholar] [CrossRef]
- Hodson, D.J.; Legros, C.; Desarménien, M.G.; Guérineau, N.C. Roles of connexins and pannexins in (neuro)endocrine physiology. Cell. Mol. Life Sci. 2015, 72, 2911–2928. [Google Scholar] [CrossRef]
- Darr, E.A.; Patel, A.D.; Yu, G.; Komorowski, Z.; McCormick, S.; Tiwari, R.; Schantz, S.P.; Geliebter, J. Reduced Cx43 gap junction plaque expression differentiates thyroid carcinomas from benign disease. Arch. Otolaryngol. Head Neck Surg. 2011, 137, 1161–1165. [Google Scholar] [CrossRef]
- Dominguez, C.; Karayan-Tapon, L.; Desurmont, T.; Gibelin, H.; Crespin, S.; Fromont, G.; Levillain, P.; Bouche, G.; Cantereau, A.; Mesnil, M.; et al. Altered expression of the gap junction protein Connexin43 Is associated with papillary thyroid carcinomas when compared with other noncancer pathologies of the thyroid. Thyroid 2011, 21, 1057–1066. [Google Scholar] [CrossRef]
- Mitašíková, M.; Lin, H.; Soukup, T.; Imanaga, I.; Tribulová, N. Diabetes and thyroid hormones affect connexin-43 and PKC-ε expression in rat heart atria. Physiol. Res. 2009, 58, 211–217. [Google Scholar] [PubMed]
- Almeida, N.A.S.; Cordeiro, A.; Machado, D.S.; Souza, L.L.; Ortiga-Carvalho, T.M.; Campos-de-Carvalho, A.C.; Wondisford, F.E.; Pazos-Moura, C.C. Connexin40 messenger ribonucleic acid is positively regulated by thyroid hormone (TH) acting in cardiac atria via the TH receptor. Endocrinology 2009, 150, 546. [Google Scholar] [CrossRef] [PubMed]
- Osuna, P.M.; Udovcic, M.; Sharma, M.D. Hyperthyroidism and the heart. Methodist Debakey Cardiovasc. J. 2017, 13, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Renaudon, B.; Lenfant, J.; Decressac, S.; Bois, P. Thyroid hormone increases the conductance density of f-channels in rabbit sino-atrial node cells. Recept. Channels 2000, 7, 1–8. [Google Scholar] [PubMed]
- Sun, Z.Q.; Ojamaa, K.; Nakamura, T.Y.; Artman, M.; Klein, I.; Coetzee, W.A. Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J. Mol. Cell. Cardiol. 2001, 33, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Awais, D.; Shao, Y.; Ismail-Beigi, F. Thyroid hormone regulation of myocardial Na/K-ATPase gene expression. J. Mol. Cell. Cardiol. 2000, 32, 1969–1980. [Google Scholar] [CrossRef]
- Shenoy, R.; Klein, I.; Ojamaa, K. Differential regulation of SR calcium transporters by thyroid hormone in rat atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, 1690–1696. [Google Scholar] [CrossRef]
- Ma, M.L.; Watanabe, K.; Watanabe, H.; Hosaka, Y.; Komura, S.; Aizawa, Y.; Yamamoto, T. Different gene expression of potassium channels by thyroid hormone and an antithyroid drug between the atrium and ventricle of rats. Jpn. Heart J. 2003, 44, 101–110. [Google Scholar] [CrossRef][Green Version]
- Hoit, B.D.; Khoury, S.F.; Shao, Y.; Gabel, M.; Liggett, S.B.; Walsh, R.A. Effects of thyroid hormone on cardiac β-adrenergic responsiveness in conscious baboons. Circulation 1997, 96, 592–598. [Google Scholar] [CrossRef]
- Morkin, E. Regulation of myosin heavy chain genes in the heart. Circulation 1993, 87, 1451–1460. [Google Scholar] [CrossRef]
- Watanabe, H.; Washizuka, T.; Komura, S.; Yoshida, T.; Hosaka, Y.; Hatada, K.; Aizawa, Y.; Chinushi, M.; Yamamoto, T.; Ma, M.; et al. Genomic and non-genomic regulation of L-type calcium channels in rat ventricle by thyroid hormone. Endocr. Res. 2005, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, M.; Yamakawa, M.; Shimabukuro, M.; Higa, N.; Takasu, N.; Kosugi, T. Electrophysiologic characteristics of atrial myocytes in levo-thyroxine-treated rats. Thyroid 2005, 15, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, Y.; Shinohara, O.; Ishida, H.; Hayashi, Y.; Nakazawa, H. Decreased protein kinase C-epsilon expression in hypertrophied cardiac ventricles induced by triiodothyronine treatment in the rat. Life Sci. 2000, 67, 1859–1868. [Google Scholar] [CrossRef]
- Connelly, T.J.; El-Hayek, R.; Sukhareva, M.; Coronado, R. L-thyroxine activates the intracellular Ca2+ release channel of skeletal muscle sarcoplasmic reticulum. Biochem. Mol. Biol. Int. 1994, 32, 441–448. [Google Scholar] [PubMed]
- Wang, Y.G.; Dedkova, E.N.; Fiening, J.P.; Ojamaa, K.; Blatter, L.A.; Lipsius, S.L. Acute exposure to thyroid hormone increases Na+ current and intracellular Ca2+ in cat atrial myocytes. J. Physiol. 2003, 546, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C.; Baumgarten, C.M. Bursting of cardiac sodium channels after acute exposure to 3,5,3′-triiodo-L-thyronine. Circ. Res. 1993, 73, 301–313. [Google Scholar] [CrossRef]
- Le Bouter, S.; Demolombe, S.; Chambellan, A.; Bellocq, C.; Aimond, F.; Toumaniantz, G.; Lande, G.; Siavoshian, S.; Baró, I.; Pond, A.L.; et al. Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: Relation to cellular and integrated electrophysiology. Circ. Res. 2003, 92, 234–242. [Google Scholar] [CrossRef]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Romanelli, M.N.; Cerbai, E. The hyperpolarization-activated cyclic nucleotide-gated channels: From biophysics to pharmacology of a unique family of ion channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Wustmann, K.; Kucera, J.P.; Zanchi, A.; Burow, A.; Stuber, T.; Chappuis, B.; Diem, P.; Delacrétaz, E. Activation of electrical triggers of atrial fibrillation in hyperthyroidism. J. Clin. Endocrinol. Metab. 2008, 93, 2104–2108. [Google Scholar] [CrossRef]
- Ehrlich, J.R.; Biliczki, P.; Hohnloser, S.H.; Nattel, S. Atrial-Selective Approaches for the Treatment of Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 787–792. [Google Scholar] [CrossRef]
- Hu, Y.; Jones, S.V.P.; Dillmann, W.H. Effects of hyperthyroidism on delayed rectifier K+ currents in left and right murine atria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 1448–1455. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Komiya, N.; Isomoto, S.; Nakao, K.; Hayano, M.; Yano, K. Electrophysiological abnormalities of the atrial muscle in patients with paroxysmal atrial fibrillation associated with hyperthyroidism. Clin. Endocrinol. 2002, 56, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Katircibasi, M.T.; Deniz, F.; Pamukcu, B.; Binici, S.; Atar, I. Effects of short-term propylthiouracil treatment on P wave duration and P wave dispersion in patients with overt hypertyroidism. Exp. Clin. Endocrinol. Diabetes 2007, 115, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Colkesen, Y.; Acil, T.; Abayli, B.; Yigit, F.; Katircibasi, T.; Kocum, T.; Demircan, S.; Sezgin, A.; Ozin, B.; Muderrisoglu, H. Mean platelet volume is elevated during paroxysmal atrial fibrillation: A marker of increased platelet activation? Blood Coagul. Fibrinolysis 2008, 19, 411. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Jones, N.R.; Taylor, C.J.; Hobbs, F.D.R.; Bowman, L.; Casadei, B. Screening for atrial fibrillation: A call for evidence. Eur. Heart J. 2020, 41, 1075–1085. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).