Para-Halogenation of Amphetamine and Methcathinone Increases the Mitochondrial Toxicity in Undifferentiated and Differentiated SH-SY5Y Cells


 , , and
, , and
Abstract
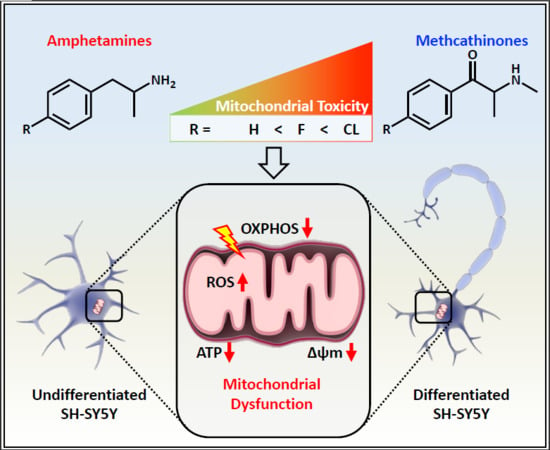
1. Introduction
2. Results
2.1. Cell Differentiation
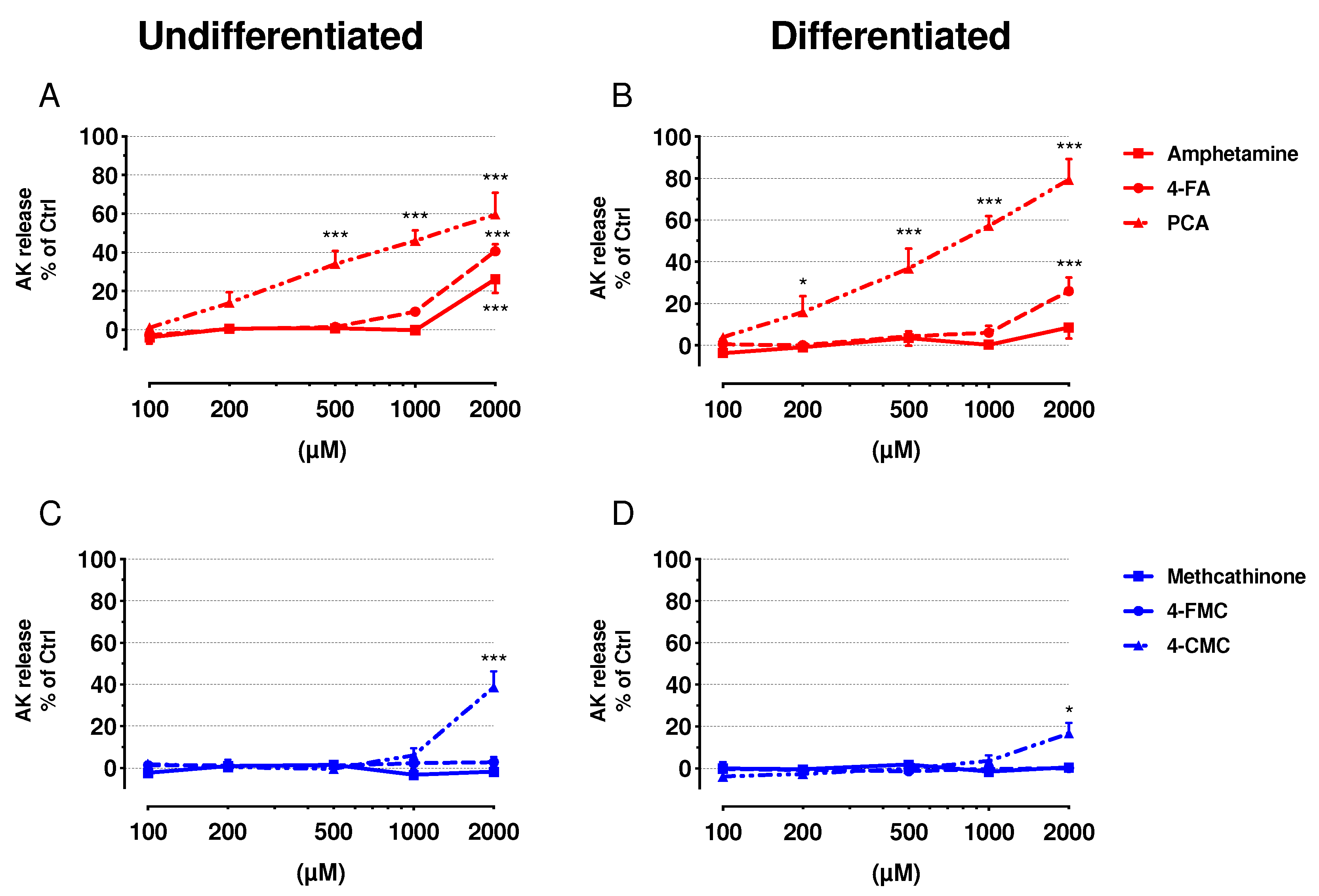
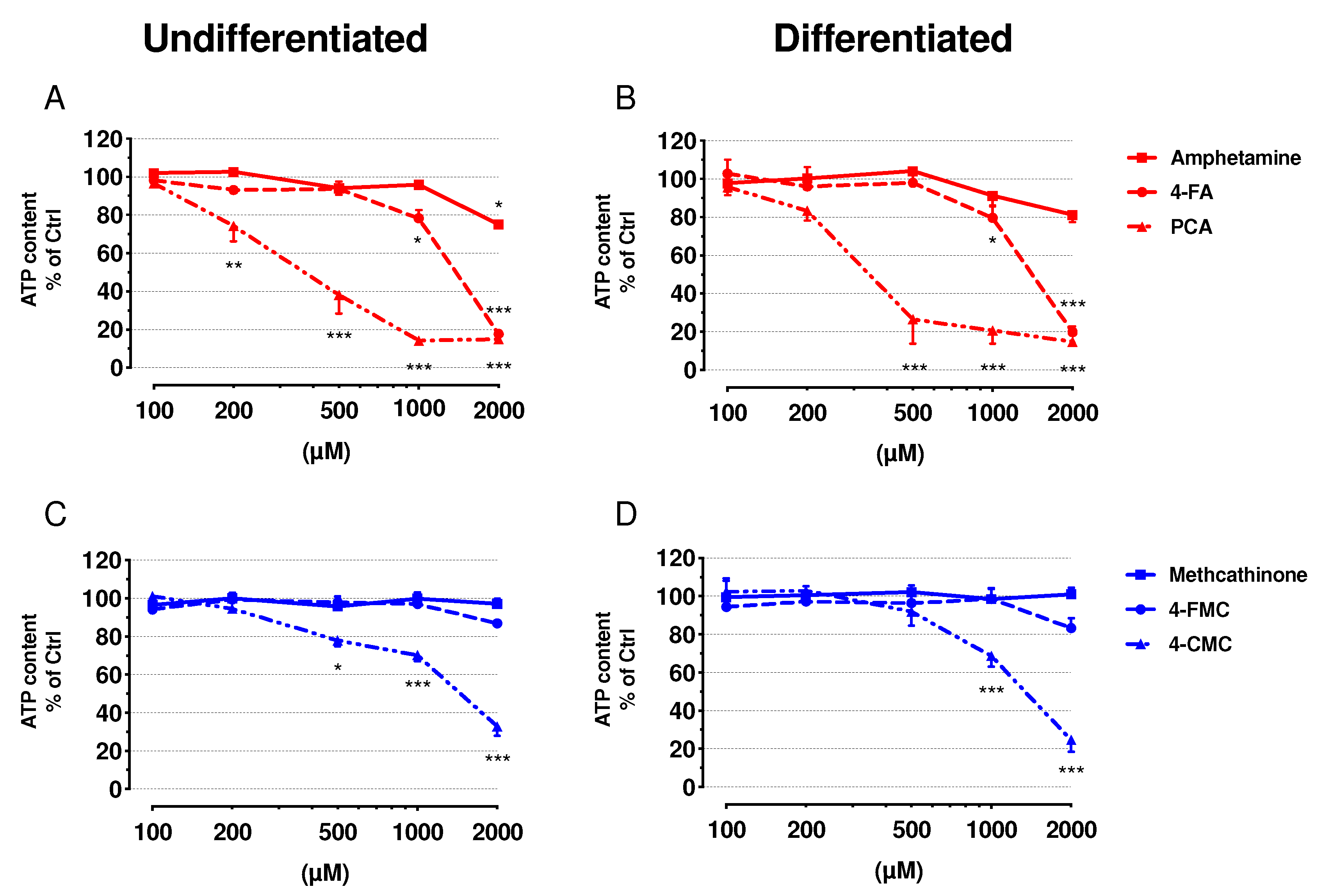
2.2. Cell Membrane Integrity and ATP Content
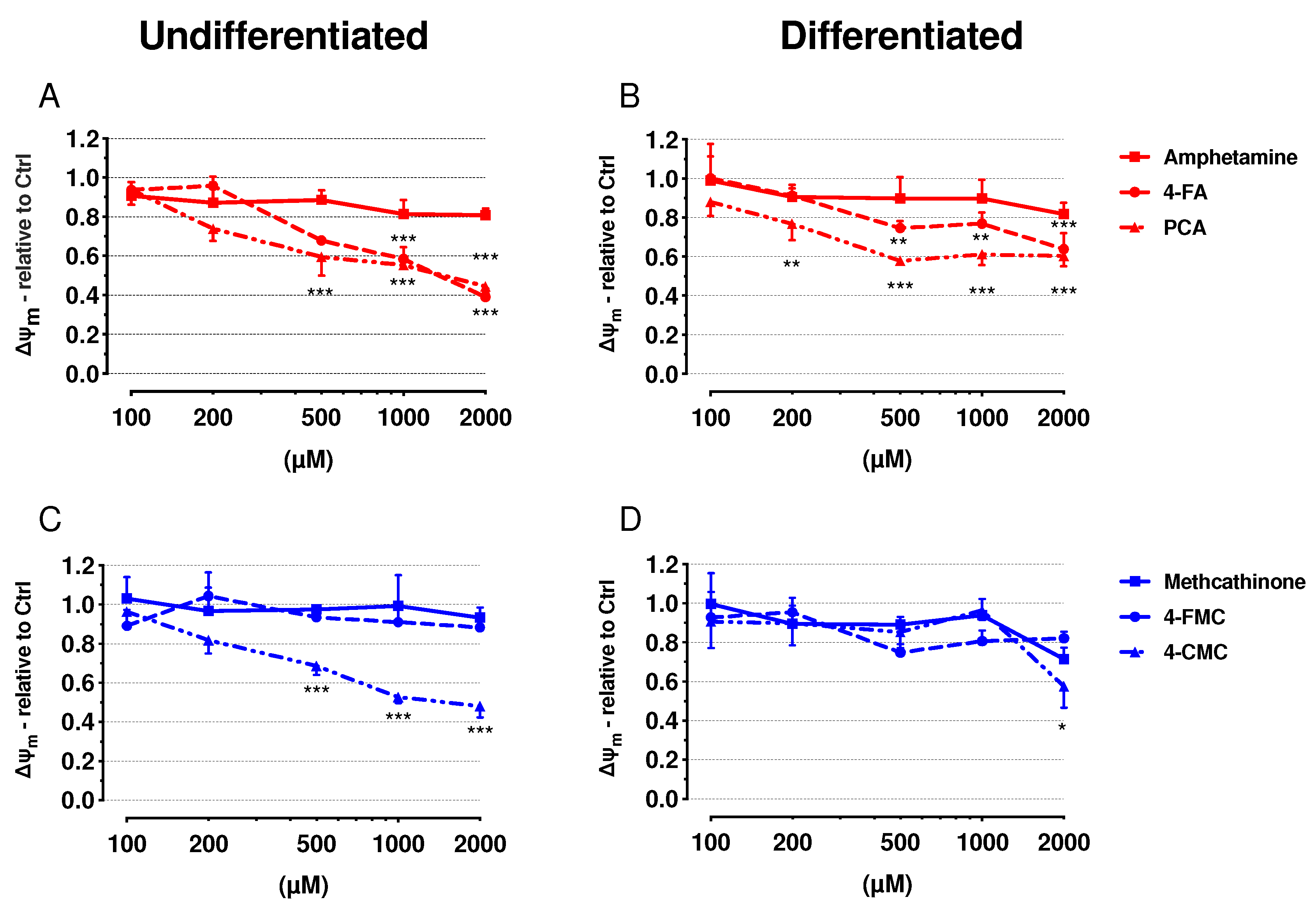
2.3. Mitochondrial Membrane Potential
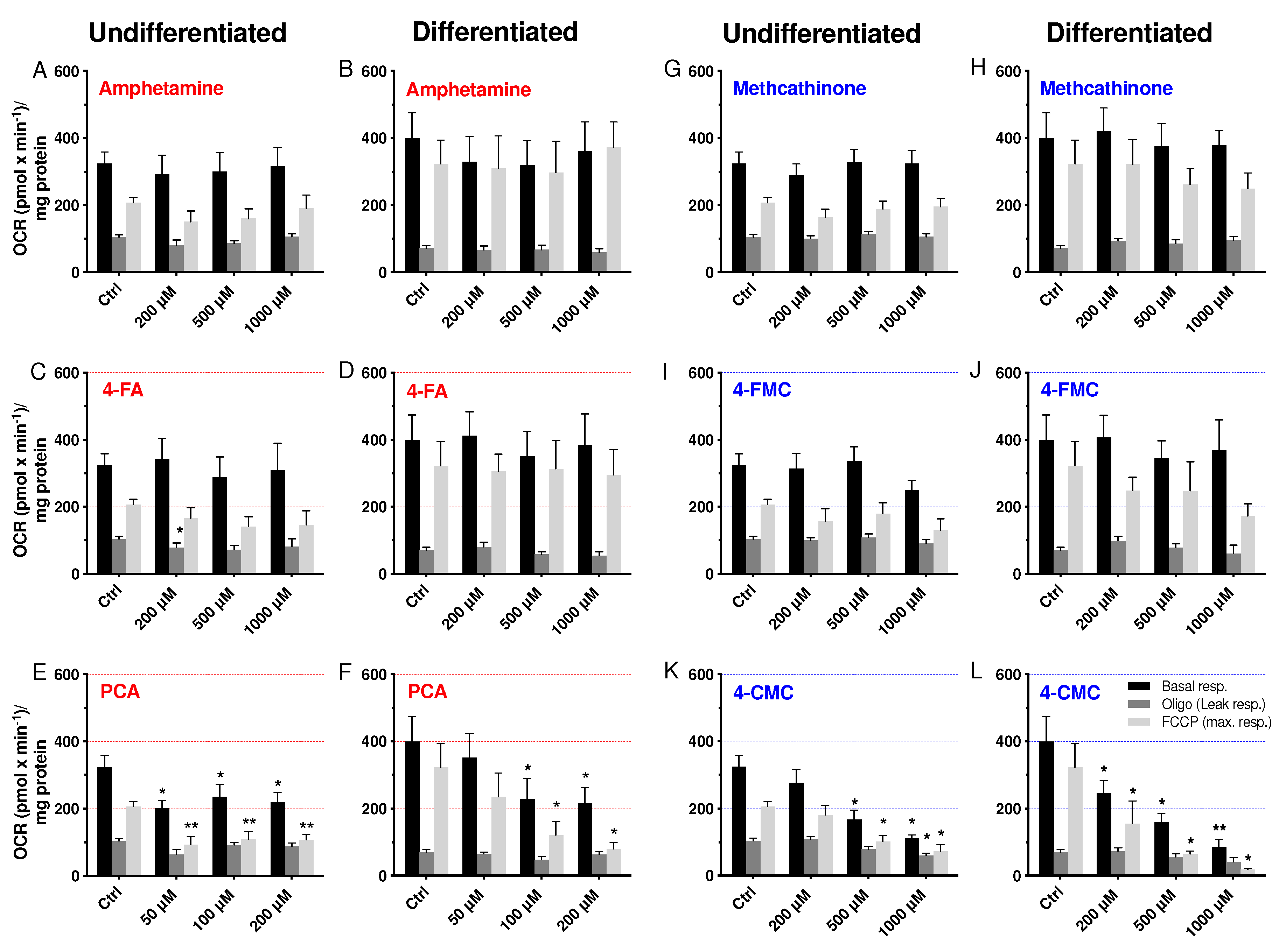
2.4. Cellular Oxygen Consumption
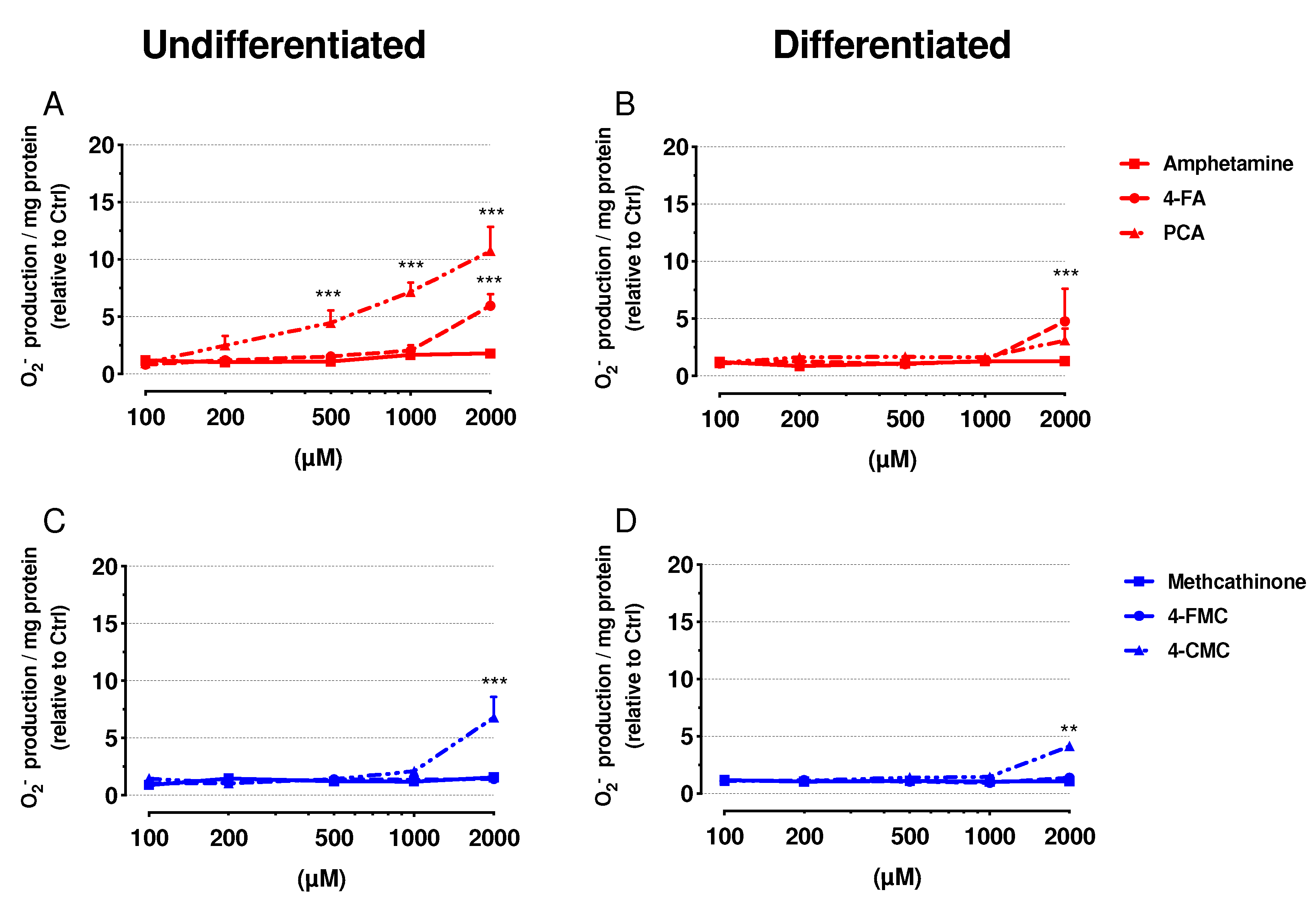
2.5. Mitochondrial Superoxide Production
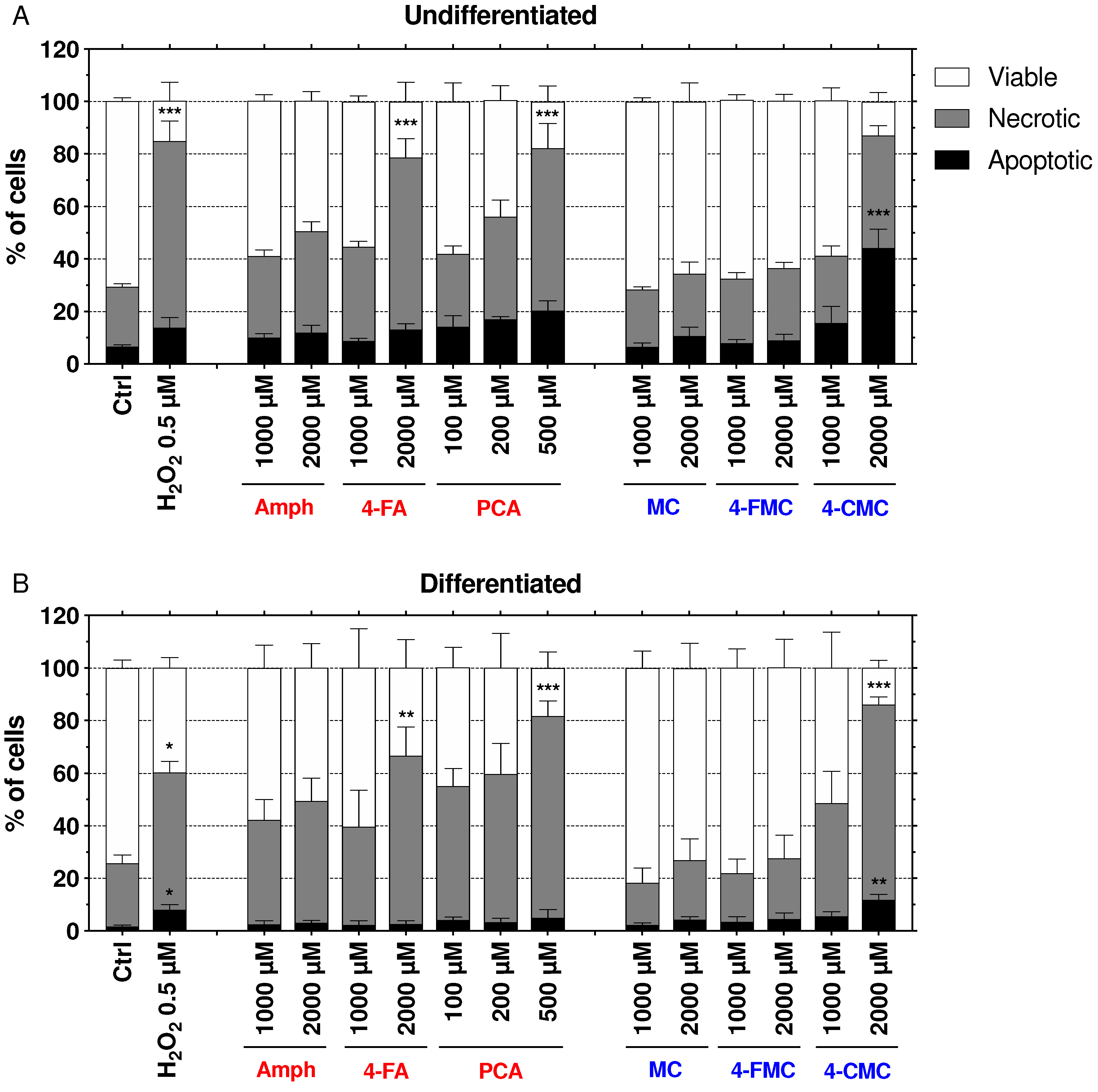
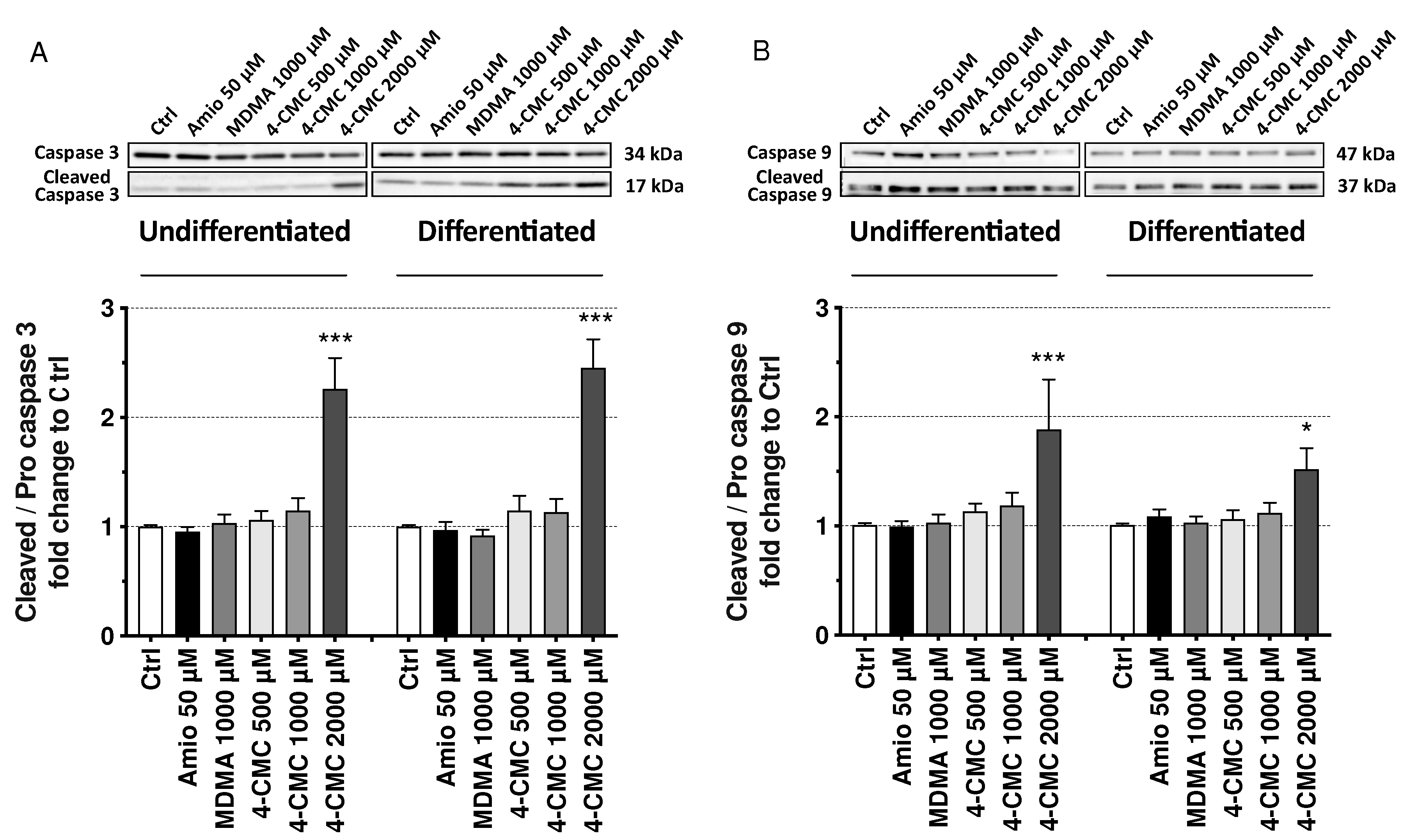
2.6. Mechanisms of Cell Death
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Differentiation
4.3. Cell Differentiation Determined by Microscopy
4.4. Cell Membrane Integrity
4.5. ATP Content
4.6. Mitochondrial Membrane Potential
4.7. Oxygen Consumption
4.8. Mitochondrial Superoxide Production
4.9. Annexin V/Propidium Iodide Staining
4.10. Western Blotting
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Heal, D.J.; Smith, S.L.; Gosden, J.; Nutt, D.J. Amphetamine, past and present-a pharmacological and clinical perspective. J. Psychopharmacol. 2013, 27, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Luethi, D.; Walter, M.; Zhou, X.; Rudin, D.; Krahenbuhl, S.; Liechti, M.E. Para-halogenation affects monoamine transporter inhibition properties and hepatocellular toxicity of amphetamines and methcathinones. Front. Pharmacol. 2019, 10, 438. [Google Scholar] [CrossRef] [PubMed]
- Calkins, R.F.; Aktan, G.B.; Hussain, K.L. Methcathinone: the next illicit stimulant epidemic? J. Psychoact. Drugs 1995, 27, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Kesha, K.; Boggs, C.L.; Ripple, M.G.; Allan, C.H.; Levine, B.; Jufer-Phipps, R.; Doyon, S.; Chi, P.; Fowler, D.R. Methylenedioxypyrovalerone ("bath salts"), related death: case report and review of the literature. J. Forensic Sci. 2013, 58, 1654–1659. [Google Scholar] [CrossRef]
- Neumann, C.S.; Fujimori, D.G.; Walsh, C.T. Halogenation strategies in natural product biosynthesis. Chem. Biol. 2008, 15, 99–109. [Google Scholar] [CrossRef]
- Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. Halogenation of drugs enhances membrane binding and permeation. Chembiochem 2004, 5, 676–684. [Google Scholar] [CrossRef]
- Brandt, S.D.; Sumnall, H.R.; Measham, F.; Cole, J. Analyses of second-generation 'legal highs' in the UK: initial findings. Drug Test. Anal. 2010, 2, 377–382. [Google Scholar] [CrossRef]
- Grifell, M.; Ventura, M.; Carbon, X.; Quintana, P.; Galindo, L.; Palma, A.; Fornis, I.; Gil, C.; Farre, M.; Torrens, M. Patterns of use and toxicity of new para-halogenated substituted cathinones: 4-CMC (clephedrone), 4-CEC (4-chloroethcatinone) and 4-BMC (brephedrone). Hum. Psychopharmacol. Clin. Exp. 2017, 32, e2621. [Google Scholar] [CrossRef]
- Linsen, F.; Koning, R.P.; van Laar, M.; Niesink, R.J.; Koeter, M.W.; Brunt, T.M. 4-Fluoroamphetamine in the Netherlands: more than a one-night stand. Addiction 2015, 110, 1138–1143. [Google Scholar] [CrossRef]
- Tomczak, E.; Wozniak, M.K.; Kata, M.; Wiergowski, M.; Szpiech, B.; Biziuk, M. Blood concentrations of a new psychoactive substance 4-chloromethcathinone (4-CMC) determined in 15 forensic cases. Forensic Toxicol. 2018, 36, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Archer, R.P. Fluoromethcathinone, a new substance of abuse. Forensic Sci. Int. 2009, 185, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Fuller, R.W. Effects ofp-chloroamphetamine on brain serotonin neurons. Neurochem. Res. 1992, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, B.K.; Moszczynska, A.; Gudelsky, G.A. Amphetamine toxicities: Classical and emerging mechanisms. Ann. New York Acad. Sci. 2010, 1187, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Lewis, O.; Kumar, R.; Yeruva, S.L.; Curry, B.H. Amphetamine abuse related acute myocardial infarction. Case Rep. Cardiol. 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.M.; Kuczenski, R.; McCracken, J.T.; London, E.D. Potential adverse effects of amphetamine treatment on brain and behavior: a review. Mol. Psychiatry 2009, 14, 123–142. [Google Scholar] [CrossRef]
- Kalix, P. The pharmacology of khat. Gen. Pharmacol. Vasc. Syst. 1984, 15, 179–187. [Google Scholar] [CrossRef]
- Luethi, D.; Liechti, M.E.; Krähenbühl, S. Mechanisms of hepatocellular toxicity associated with new psychoactive synthetic cathinones. Toxicology 2017, 387, 57–66. [Google Scholar] [CrossRef]
- Zhou, X.; Luethi, D.; Sanvee, G.M.; Bouitbir, J.; Liechti, M.E.; Krahenbuhl, S. Molecular Toxicological Mechanisms of Synthetic Cathinones on C2C12 Myoblasts. Int. J. Mol. Sci. 2019, 20, 1561. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V.; Gallego, C.; Comella, J.X. Sequential treatment of sh-sy5y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef]
- Agholme, L.; Lindstrom, T.; Kagedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimer's Dis. 2010, 20, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Ryu, P.D.; Lee, S.Y. Kv3.4 is modulated by HIF-1α to protect SH-SY5Y cells against oxidative stress-induced neural cell death. Sci. Rep. 2017, 7, 2075. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Apoptosis and necrosis: Intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997, 4, 429. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-M.; Yang, C.-Q.; Cheng, B.-H.; Chen, J.; Bai, B. Orexin-A protects SH-SY5Y cells against H2O2-induced oxidative damage via the PI3K/MEK1/2/ERK1/2 signaling pathway. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418785739. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Nielsen, I.M.; Dubnick, B. Amphetamines and Related Compounds. In Proceedings of the Mario Negri Institute for Pharmacological Research; Raven Press: New York, NY, USA, 1970; p. 63. [Google Scholar]
- Barbosa, D.J.; Capela, J.P.; Feio-Azevedo, R.; Teixeira-Gomes, A.; Bastos Mde, L.; Carvalho, F. Mitochondria: Key players in the neurotoxic effects of amphetamines. Arch. Toxicol. 2015, 89, 1695–1725. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; Rago, J.-P.D.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. Embo J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Colado, M.I.; Murray, T.K.; Green, A.R. 5-HT loss in rat brain following 3,4-methylenedioxymethamphetamine (MDMA), p-chloroamphetamine and fenfluramine administration and effects of chlormethiazole and dizocilpine. Br. J. Pharmacol. 1993, 108, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.J.; Anderholm, D.C.; Ames, M.M. Metabolic activation of the serotonergic neurotoxin para-chloroamphetamine to chemically reactive intermediates by hepatic and brain microsomal preparations. Biochem Pharm. 1986, 35, 1737–1742. [Google Scholar] [CrossRef]
- Hyun, D.H.; Hunt, N.D.; Emerson, S.S.; Hernandez, J.O.; Mattson, M.P.; Cabo, R.D. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J. Neurochem. 2007, 100, 1364–1374. [Google Scholar] [CrossRef]
- Volobueva, A.S.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Mitochondrial genome variability: the effect on cellular functional activity. Ther. Clin. Risk Manag. 2018, 14, 237–245. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Halpin, L.E.; Collins, S.A.; Yamamoto, B.K. Neurotoxicity of methamphetamine and 3, 4-methylenedioxymethamphetamine. Life Sci. 2014, 97, 37–44. [Google Scholar] [CrossRef]
- Carvalho, M.; Carmo, H.; Costa, V.M.; Capela, J.P.; Pontes, H.; Remião, F.; Carvalho, F.; de Lourdes Bastos, M. Toxicity of amphetamines: an update. Arch. Toxicol. 2012, 86, 1167–1231. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Bankson, M.G. Amphetamine neurotoxicity: cause and consequence of oxidative stress. Crit. Rev. Neurobiol. 2005, 17. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.J.o.; Bastos, M.d.L.; Fernandes, E.; Carvalho, F.l.; Guedes de Pinho, P.; Carvalho, M.R. Neurotoxicity of β-keto amphetamines: deathly mechanisms elicited by methylone and MDPV in human dopaminergic SH-SY5Y cells. ACS Chem. Neurosci. 2017, 8, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Rizzuto, R. Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 2010, 131, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Naserzadeh, P.; Taghizadeh, G.; Atabaki, B.; Seydi, E.; Pourahmad, J. A comparison of mitochondrial toxicity of mephedrone on three separate parts of brain including hippocampus, cortex and cerebellum. Neurotoxicology 2019, 73, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Roos, N.J.; Duthaler, U.; Bouitbir, J.; Krahenbuhl, S. The uricosuric benzbromarone disturbs the mitochondrial redox homeostasis and activates the NRF2 signaling pathway in HepG2 cells. Free Radic. Biol. Med. 2020, 152, 216–226. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Zhou, X.; Bouitbir, J.; Liechti, M.E.; Krähenbühl, S.; Mancuso, R.V. Hyperthermia Increases Neurotoxicity Associated with Novel Methcathinones. Cells 2020, 9, 965. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts:Requirement for datp and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Schneider, L.; Giordano, S.; Zelickson, B.R.; Johnson, M.S.; Benavides, G.A.; Ouyang, X.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic. Biol. Med. 2011, 51, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.T.; Lau, W.K.W.; Yu, M.S.; Lai, C.S.W.; Yeung, S.C.; So, K.F.; Chang, R.C.C. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 2009, 30, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Lappin, J.M.; Darke, S.; Farrell, M. Methamphetamine use and future risk for Parkinson’s disease: Evidence and clinical implications. Drug Alcohol Depend. 2018, 187, 134–140. [Google Scholar] [CrossRef]
- Lopes, F.M.; Schröder, R.; da Frota Júnior, M.L.C.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F. Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Dolder, P.C.; Strajhar, P.; Vizeli, P.; Hammann, F.; Odermatt, A.; Liechti, M.E. Pharmacokinetics and Pharmacodynamics of Lisdexamfetamine Compared with D-Amphetamine in Healthy Subjects. Front. Pharmacol. 2017, 8, 617. [Google Scholar] [CrossRef] [PubMed]
- Gerets, H.H.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef]
- Valente, M.J.; Araujo, A.M.; Silva, R.; Bastos Mde, L.; Carvalho, F.; Guedes de Pinho, P.; Carvalho, M. 3,4-Methylenedioxypyrovalerone (MDPV): in vitro mechanisms of hepatotoxicity under normothermic and hyperthermic conditions. Arch. Toxicol. 2016, 90, 1959–1973. [Google Scholar] [CrossRef]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y human neuroblastoma cell line. J. Vis. Exp. 2016, 108, 53193. [Google Scholar] [CrossRef]
- Haylett, W.; Swart, C.; van der Westhuizen, F.; van Dyk, H.; van der Merwe, L.; van der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered mitochondrial respiration and other features of mitochondrial function in parkin-mutant fibroblasts from parkinson's disease patients. Parkinson’s Dis. 2016, 2016, 1819209. [Google Scholar] [CrossRef]
- Felser, A.; Blum, K.; Lindinger, P.W.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of hepatocellular toxicity associated with dronedarone-a comparison to amiodarone. Toxicol. Sci. 2013, 131, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. Beyond annexin V: Fluorescence response of cellular membranes to apoptosis. Cytotechnology 2013, 65, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, D.J.; Capela, J.P.; Silva, R.; Ferreira, L.M.; Branco, P.S.; Fernandes, E.; Bastos, M.L.; Carvalho, F. "Ecstasy"-induced toxicity in SH-SY5Y differentiated cells: role of hyperthermia and metabolites. Arch. Toxicol. 2014, 88, 515–531. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Amph | 4-FA | PCA | MC | 4-FMC | 4-CMC | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell | und | diff | und | diff | und | diff | und | diff | und | diff | und | diff |
| MT (IC50) [mM] | >2 | >2 | >2 | >2 | 1.17 | 0.77 | >2 | >2 | >2 | >2 | >2 | >2 |
| ATP (IC50) [mM] | >2 | >2 | 1.44 | 1.41 | 0.42 | 0.39 | >2 | >2 | >2 | >2 | 1.43 | 1.33 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Bouitbir, J.; Liechti, M.E.; Krähenbühl, S.; Mancuso, R.V. Para-Halogenation of Amphetamine and Methcathinone Increases the Mitochondrial Toxicity in Undifferentiated and Differentiated SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 2841. https://doi.org/10.3390/ijms21082841
Zhou X, Bouitbir J, Liechti ME, Krähenbühl S, Mancuso RV. Para-Halogenation of Amphetamine and Methcathinone Increases the Mitochondrial Toxicity in Undifferentiated and Differentiated SH-SY5Y Cells. International Journal of Molecular Sciences. 2020; 21(8):2841. https://doi.org/10.3390/ijms21082841
Chicago/Turabian StyleZhou, Xun, Jamal Bouitbir, Matthias E. Liechti, Stephan Krähenbühl, and Riccardo V. Mancuso. 2020. "Para-Halogenation of Amphetamine and Methcathinone Increases the Mitochondrial Toxicity in Undifferentiated and Differentiated SH-SY5Y Cells" International Journal of Molecular Sciences 21, no. 8: 2841. https://doi.org/10.3390/ijms21082841
APA StyleZhou, X., Bouitbir, J., Liechti, M. E., Krähenbühl, S., & Mancuso, R. V. (2020). Para-Halogenation of Amphetamine and Methcathinone Increases the Mitochondrial Toxicity in Undifferentiated and Differentiated SH-SY5Y Cells. International Journal of Molecular Sciences, 21(8), 2841. https://doi.org/10.3390/ijms21082841