Current Symptomatic and Disease-Modifying Treatments in Multiple System Atrophy

Abstract
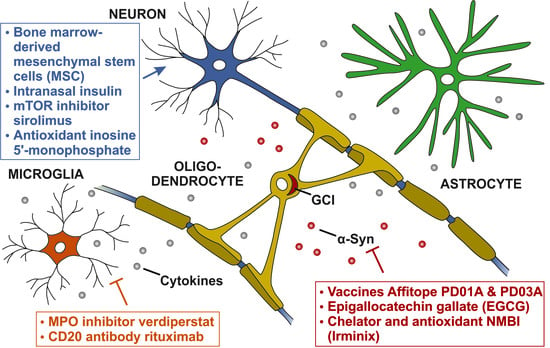
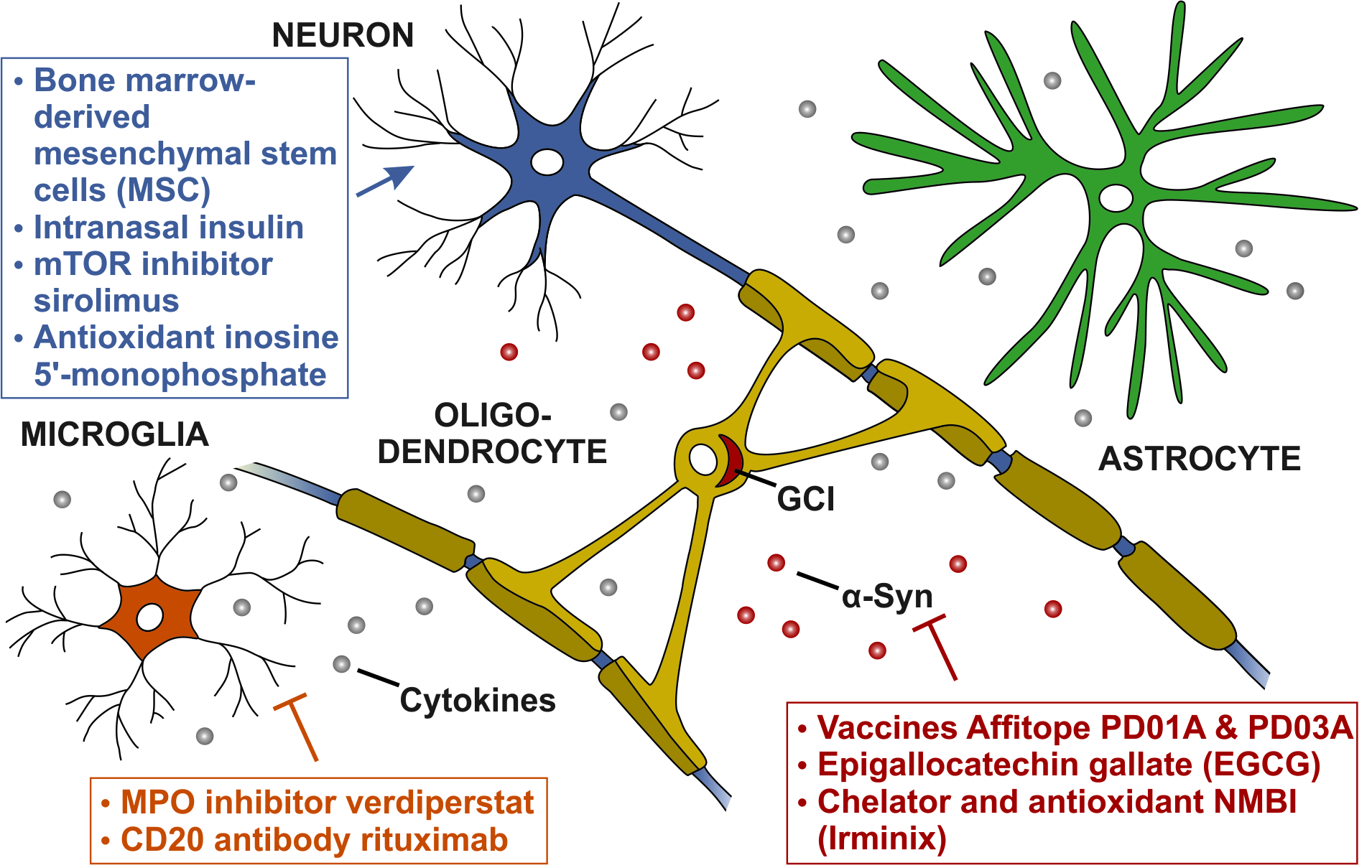
1. Introduction
2. Neuropathology of MSA
3. Current Symptomatic Treatment of MSA
4. Potential Disease-Modifying Targets in MSA
4.1. Targeting alpha-Synuclein Aggregation
4.2. Targeting Neuroinflammation
4.3. Targeting Neuronal Loss
5. Limitations
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-NP | 3-nitropropionic acid |
| AD | Alzheimer’s disease |
| BDNF | brain-derived neurotrophic factor |
| CGI-C | Clinical Global Impression of Change |
| CNP | 2′-3′-cyclic nucleotide 3′-phosphodiesterase |
| CNP-hα-syn mice | transgenic MSA mouse model overexpressing α-syn under a 2′-3′-cyclic nucleotide 3′-phosphodiesterase promoter |
| CNS | central nervous system |
| EAAT2 | excitatory amino acid transporter 2 |
| EGCG | epigallocatechin gallate |
| GCI | glial cytoplasmic inclusion |
| GDH2 | glutamate dehydrogenase |
| GDNF | glial cell line-derived neurotrophic factor |
| hA53T α-syn | human A53T mutant form of α-syn |
| IL | interleukins |
| LRRK2 | leucine rich repeat kinase 2 |
| MAO-B | monoaminooxidase B |
| MBP | myelin basic protein |
| MBP1-hα-syn mice | transgenic MSA mouse model expressing human α-syn under control of the myelin basic protein line 1 |
| MPO | myeloperoxidase |
| MS | multiple sclerosis |
| MSA | multiple system atrophy |
| MSA-C | cerebellar subtype of MSA |
| MSA-P | parkinsonian subtype of MSA |
| MSA-QoL | MSA-Quality of Life |
| MSCs | mesenchymal stem cells |
| mTOR | mechanistic target of rapamycin |
| n.a. | not applicable |
| NET | norepinephrine transporter |
| NRF2 | nuclear-related factor 2 |
| NRI | norepinephrine reuptake inhibitor |
| NMDA | N-methyl-d- aspartic acid |
| PD | Parkinson’s disease |
| PDGFβ | platelet-derived growth factor β |
| PLP-hα-syn mice | transgenic MSA mouse model overexpressing human α-syn under the proteolipid protein promoter |
| PSP | progressive supranuclear palsy |
| TNFα | tumor necrosis factor alpha |
| TPPP/p25a | tubulin polymerization-promoting protein |
| UMSARS | Unified Multiple System Atrophy Rating Scale |
| VEGF | vascular endothelial growth factor |
| α-syn | alpha-synuclein |
References
- Wenning, G.K.; Colosimo, C.; Geser, F.; Poewe, W. Multiple system atrophy. Lancet Neurol. 2004, 3, 93–103. [Google Scholar] [CrossRef]
- Gilman, S.; Wenning, G.; Low, P.A.; Brooks, D.; Mathias, C.; Trojanowski, J.; Wood, N.W.; Colosimo, C.; Dürr, A.; Fowler, C. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.I.; Kahn, J.E.; Lantos, P.L. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J. Neurol. Sci. 1989, 94, 79–100. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Burré, J. The Synaptic Function of alpha-Synuclein. J. Parkinson’s Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.S.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: An In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Reports 2015, 5, 174–184. [Google Scholar] [CrossRef]
- Kisos, H.; Pukaß, K.; Ben-Hur, T.; Richter-Landsberg, C.; Sharon, R. Increased Neuronal α-Synuclein Pathology Associates with Its Accumulation in Oligodendrocytes in Mice Modeling α-Synucleinopathies. PLoS ONE 2012, 7, e46817. [Google Scholar] [CrossRef]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.C.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases α-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef]
- Hasegawa, T.; Baba, T.; Kobayashi, M.; Konno, M.; Sugeno, N.; Kikuchi, A.; Itoyama, Y.; Takeda, A. Role of TPPP/p25 on α-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy☆. Neurochem. Int. 2010, 57, 857–866. [Google Scholar] [CrossRef]
- Ahmed, Z.; Asi, Y.T.; Lees, A.J.; Revesz, T.; Holton, J.L. Identification and Quantification of Oligodendrocyte Precursor Cells in Multiple System Atrophy, Progressive Supranuclear Palsy and Parkinson’s Disease. Brain Pathol. 2013, 23, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Ettle, B.; Kerman, B.E.; Valera, E.; Gillmann, C.; Schlachetzki, J.C.M.; Reiprich, S.; Büttner, C.; Ekici, A.B.; Reis, A.; Wegner, M.; et al. alpha-Synuclein-induced myelination deficit defines a novel interventional target for multiple system atrophy. Acta Neuropathol. 2016, 132, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, I.; Giasson, B.I.; Sasaki, R.; Zhang, B.; Joyce, S.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M.-Y. Mouse Model of Multiple System Atrophy α-Synuclein Expression in Oligodendrocytes Causes Glial and Neuronal Degeneration. Neuron 2005, 45, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, A.; Akiguchi, I.; Lee, G.C.; McGeer, E.G.; McGeer, P.L.; Kimura, J. Myelin Degeneration in Multiple System Atrophy Detected by Unique Antibodies. Am. J. Pathol. 1998, 153, 735–744. [Google Scholar] [CrossRef]
- Ozawa, T. Morphological substrate of autonomic failure and neurohormonal dysfunction in multiple system atrophy: Impact on determining phenotype spectrum. Acta Neuropathol. 2007, 114, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, L.; Ullerup, B.H.; Sunay, F.B.; Brudek, T.; Løkkegaard, A.; Agander, T.K.; Winge, K.; Pakkenberg, B. Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy — A stereological study. Neurobiol. Dis. 2015, 74, 104–113. [Google Scholar] [CrossRef]
- May, V.E.L.; Ettle, B.; Poehler, A.-M.; Nuber, S.; Ubhi, K.; Rockenstein, E.; Winner, B.; Wegner, M.; Masliah, E.; Winkler, J. alpha-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol. Aging 2014, 35, 2357–2368. [Google Scholar] [CrossRef]
- Salvesen, L.; Winge, K.; Brudek, T.; Agander, T.K.; Løkkegaard, A.; Pakkenberg, B. Neocortical Neuronal Loss in Patients with Multiple System Atrophy: A Stereological Study. Cereb. Cortex 2015, 27, 400–410. [Google Scholar] [CrossRef][Green Version]
- Yoshida, M. Multiple system atrophy: alpha-synuclein and neuronal degeneration. Neuropathology 2007, 27, 484–493. [Google Scholar] [CrossRef]
- Vieira, B.D.M.; Radford, R.A.; Chung, R.; Guillemin, G.J.; Pountney, D. Neuroinflammation in Multiple System Atrophy: Response to and Cause of α-Synuclein Aggregation. Front. Cell. Neurosci. 2015, 9, 437. [Google Scholar] [CrossRef]
- Radford, R.; Rcom-H’Cheo-Gauthier, A.; Wong, M.; Eaton, E.D.; Quilty, M.; Blizzard, C.A.; Norazit, A.; Meedeniya, A.; Vickers, J.C.; Gai, W.P.; et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α-synuclein inclusions. Mol. Cell. Neurosci. 2015, 65, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Ettle, B.; Battis, K.; Reiprich, S.; Schlachetzki, J.C.M.; Masliah, E.; Wegner, M.; Kuhlmann, T.; Riemenschneider, M.J.; Winkler, J. Oligodendroglial alpha-synucleinopathy-driven neuroinflammation in multiple system atrophy. Brain Pathol. 2019, 29, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Collaboration M.-S.A.R. Mutations in COQ2 in Familial and Sporadic Multiple-System Atrophy. N. Engl. J. Med. 2013, 369, 233–244. [Google Scholar]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Sailer, A.; Scholz, S.W.; Nalls, M.A.; Schulte, C.; Federoff, M.; Price, T.R.; Lees, A.; Ross, O.A.; Dickson, D.W.; Mok, K.; et al. A genome-wide association study in multiple system atrophy. Neurology 2016, 87, 1591–1598. [Google Scholar] [CrossRef]
- Scholz, S.W.; Houlden, H.; Schulte, C.; Sharma, M.; Li, A.; Berg, D.; Melchers, A.; Paudel, R.; Gibbs, J.R.; Simon-Sanchez, J. SNCA variants are associated with increased risk for multiple system atrophy. Ann. Neurol.: Off. J. Am. Neurol. Assoc Child Neurol. Soc. 2009, 65, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Vilarino-Guell, C.; Wszolek, Z.K.; Farrer, M.J.; Dickson, D.W. Reply to: SNCA variants are associated with increased risk of multiple system atrophy. Ann. Neurol. 2010, 67, 414. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Dürr, A.; Wood, N.; Parkinson, M.H.; Camuzat, A.; Hulot, J.-S.; Morrison, K.E.; Renton, A.E.; Süssmuth, S.D.; Landwehrmeyer, G.B.; et al. Genetic Variants of the α-Synuclein Gene SNCA Are Associated with Multiple System Atrophy. PLoS ONE 2009, 4, e7114. [Google Scholar] [CrossRef]
- Kiely, A.P.; Ling, H.; Asi, Y.T.; Kara, E.; Proukakis, C.; Schapira, A.H.V.; Morrison, K.E.; Roberts, H.C.; Lubbe, S.; Limousin, P.; et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol. Neurodegener. 2015, 10, 41. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Nee, L.E.; Gomez, M.R.; Dambrosia, J.; Bale, S.; Eldridge, R.; Polinsky, R.J. Environmental— Occupational risk factors and familial associations in multiple system atrophy: A preliminary investigation. Clin. Auton. Res. 1991, 1, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.A.; Jankovic, J.; Kirkpatrick, J.B. Multiple system atrophy: The putative causative role of environmental toxins. Arch. Neurol. 1999, 56, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.I.; Lantos, P.L. Accumulation of tubular structures in oligodendroglial and neuronal cells as the basic alteration in multiple system atrophy. J. Neurol. Sci. 1992, 107, 172–182. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Lantos, P.L. Papp–Lantos inclusions and the pathogenesis of multiple system atrophy: An update. Acta Neuropathol. 2010, 119, 657–667. [Google Scholar] [CrossRef]
- Nakamura, K.; Mori, F.; Kon, T.; Tanji, K.; Miki, Y.; Tomiyama, M.; Kurotaki, H.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; et al. Accumulation of phosphorylated α-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology 2016, 36, 157–167. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Duda, J.E.; Lee, V.M.-Y.; Trojanowski, J.Q. Neuropathology of synuclein aggregates: New insights into mechanisms of neurodegenerative diseases. J. Neurosci. Res. 2000, 61, 121–127. [Google Scholar] [CrossRef]
- Ahmed, Z.; Asi, Y.T.; Sailer, A.; Lees, A.J.; Houlden, H.; Revesz, T.; Holton, J.L. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol. Appl. Neurobiol. 2012, 38, 4–24. [Google Scholar] [CrossRef]
- Wenning, G.K.; Tison, F.; Ben Shlomo, Y.; Daniel, S.E.; Quinn, N.P. Multiple system atrophy: A review of 203 pathologically proven cases. Mov. Disord. 1997, 12, 133–147. [Google Scholar] [CrossRef]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.I.; Lantos, P.L. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 1994, 117, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Paviour, D.; Quinn, N.; Josephs, K.A.; Sangha, H.; Kilford, L.; Healy, D.G.; Wood, N.; Lees, A.J.; Holton, J.L.; et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: Clinicopathological correlations. Brain 2004, 127, 2657–2671. [Google Scholar] [CrossRef]
- Ozawa, T.; Kakita, A.; Onodera, O.; Tada, M.; Ishihara, T.; Morita, T.; Shimohata, T.; Wakabayashi, K.; Takahashi, H.; Nishizawa, M. The phenotype spectrum of Japanese multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Saito, Y.; Terao, S.; Ando, T.; Kachi, T.; Mukai, E.; Aiba, I.; Abe, Y.; Tamakoshi, A.; Doyu, M.; et al. Progression and prognosis in multiple system atrophy: An analysis of 230 Japanese patients. Brain 2002, 125, 1070–1083. [Google Scholar] [CrossRef]
- Wenning, G.K.; Stefanova, N.; Jellinger, K.A.; Poewe, W.; Schlossmacher, M.G. Multiple system atrophy: A primary oligodendrogliopathy. Ann. Neurol. 2008, 64, 239–246. [Google Scholar] [CrossRef]
- Ishizawa, K.; Komori, T.; Arai, N.; Mizutani, T.; Hirose, T. Glial cytoplasmic inclusions and tissue injury in multiple system atrophy: A quantitative study in white matter (olivopontocerebellar system) and gray matter (nigrostriatal system). Neuropathology 2008, 28, 249–257. [Google Scholar] [CrossRef]
- Ishizawa, K.; Komori, T.; Sasaki, S.; Arai, N.; Mizutani, T.; Hirose, T. Microglial Activation Parallels System Degeneration in Multiple System Atrophy. J. Neuropathol. Exp. Neurol. 2004, 63, 43–52. [Google Scholar] [CrossRef]
- Williams, G.P.; Marmion, D.J.; Schonhoff, A.M.; Jurkuvenaite, A.; Won, W.-J.; Standaert, D.G.; Kordower, J.H.; Harms, A. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol. 2020, 127, 205–212. [Google Scholar] [CrossRef]
- Safinamide for Multiple System Atrophy (MSA). Available online: https://clinicaltrials.gov/ct2/show/NCT03753763 (accessed on 20 February 2020).
- Tllsh2910 for Ataxia and Gut Microbiota Alteration in Patients of Multiple System Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT03901638 (accessed on 20 February 2020).
- Effects of Midodrine and Droxidopa on Splanchnic Capacitance in Autonomic Failure. Available online: https://clinicaltrials.gov/ct2/show/NCT02897063 (accessed on 20 February 2020).
- Sustained Effect of Droxidopa in Symptomatic Neurogenic Orthostatic Hypotension (RESTORE). Available online: https://clinicaltrials.gov/ct2/show/NCT02586623 (accessed on 20 February 2020).
- Norepinephrine Transporter Blockade, Autonomic Failure. Available online: https://clinicaltrials.gov/ct2/show/NCT02796209 (accessed on 20 February 2020).
- Clinical Effect of Ampreloxetine (TD-9855) for Treating snOH in Subjects with Primary Autonomic Failure (Sequoia Study). Available online: https://clinicaltrials.gov/ct2/show/NCT03750552 (accessed on 20 February 2020).
- Köllensperger, M.; Geser, F.; Ndayisaba, J.-P.; Boesch, S.; Seppi, K.; Ostergaard, K.; Dupont, E.; Cardozo, A.; Tolosa, E.; Abele, M.; et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: Final analysis of the European multiple system atrophy registry. Mov. Disord. 2010, 25, 2604–2612. [Google Scholar] [CrossRef]
- Wenning, G.K.; Ben Shlomo, Y.; Magalhães, M.; Daniel, S.E.; Quinn, N.P. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994, 117, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Low, P.A.; Reich, S.G.; Jankovic, J.; Shults, C.W.; Stern, M.B.; Novak, P.; Tanner, C.M.; Gilman, S.; Marshall, F.J.; Wooten, F.; et al. Natural history of multiple system atrophy in the USA: A prospective cohort study. Lancet Neurol. 2015, 14, 710–719. [Google Scholar] [CrossRef]
- Wenning, G.K.; Geser, F.; Krismer, F.; Seppi, K.; Duerr, S.; Boesch, S.; Köllensperger, M.; Goebel, G.; Pfeiffer, K.P.; Barone, P.; et al. The natural history of multiple system atrophy: A prospective European cohort study. Lancet Neurol. 2013, 12, 264–274. [Google Scholar] [CrossRef]
- Colosimo, C.; Merello, M.; Pontieri, F.E. Amantadine in parkinsonian patients unresponsive to levodopa: A pilot study. J. Neurol. 1996, 243, 422–425. [Google Scholar] [CrossRef]
- Rajrut, A.; Uitti, R.; Fenton, M.; George, D. Amantadine effectiveness in multiple system atrophy and progressive supranuclear palsy. Park. Relat. Disord. 1997, 3, 211–214. [Google Scholar] [CrossRef]
- Friedberg, A.; Erikh, I.; Nassar, M.; Sprecher, E.; Schlesinger, I. Efficacy of Parenteral Amantadine Therapy in the Treatment of Multiple System Atrophy with Predominant Parkinsonism. Clin. Neuropharmacol. 2018, 41, 160–163. [Google Scholar] [CrossRef]
- Wenning, G.K. Placebo-Controlled Trial of Amantadine in Multiple-System Atrophy. Clin. Neuropharmacol. 2005, 28, 225–227. [Google Scholar] [CrossRef]
- Ilg, W.; Bastian, A.J.; Boesch, S.; Burciu, R.G.; Celnik, P.; Claaßen, J.; Feil, K.; Kalla, R.; Miyai, I.; Nachbauer, W.; et al. Consensus paper: Management of degenerative cerebellar disorders. Cerebellum 2014, 13, 248–268. [Google Scholar] [CrossRef]
- Sanchez-Perez, A.M.; Llansola, M.; Cauli, O.; Felipo, V. Modulation of NMDA receptors in the cerebellum. II. Signaling pathways and physiological modulators regulating NMDA receptor function. Cerebellum 2005, 4, 162–170. [Google Scholar] [CrossRef]
- Müller, J.; Wenning, G.K.; Wissel, J.; Seppi, K.; Poewe, W. Botulinum toxin treatment in atypical parkinsonian disorders associated with disabling focal dystonia. J. Neurol. 2002, 249, 300–304. [Google Scholar] [CrossRef]
- Thobois, S.; Broussolle, E.; Toureille, L.; Vial, C. Severe dysphagia after botulinum toxin injection for cervical dystonia in multiple system atrophy. Mov. Disord. 2001, 16, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Halaska, M.; Ralph, G.; Wiedemann, A.; Primus, G.; Ballering-Brühl, B.; Höfner, K.; Jonas, U. Controlled, double-blind, multicentre clinical trial to investigate long-term tolerability and efficacy of trospium chloride in patients with detrusor instability. World J. Urol. 2003, 20, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Sakakibara, R.; Yasuda, K.; Yamamoto, T.; Uchiyama, T.; Liu, Z.; Yamanishi, T.; Awa, Y.; Yamamoto, K.; Hattori, T. Incomplete emptying and urinary retention in multiple-system atrophy: When does it occur and how do we manage it? Mov. Disord. 2006, 21, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, R.; Matsuda, S.; Uchiyama, T.; Yoshiyama, M.; Yamanishi, T.; Hattori, T. The effect of intranasal desmopressin on nocturnal waking in urination in multiple system atrophy patients with nocturnal polyuria. Clin. Auton. Res. 2003, 13, 106–108. [Google Scholar] [CrossRef]
- Brooks, D.J.; Redmond, S.; Mathias, C.J.; Bannister, R.; Symon, L. The effect of orthostatic hypotension on cerebral blood flow and middle cerebral artery velocity in autonomic failure, with observations on the action of ephedrine. J. Neurol. Neurosurg. Psychiatry 1989, 52, 962–966. [Google Scholar] [CrossRef]
- Jankovic, J.; Gilden, J.L.; Hiner, B.C.; Kaufmann, H.; Brown, D.C.; Coghlan, C.H.; Rubin, M.; Fouad-Tarazi, F.M. Neurogenic orthostatic hypotension: A double-blind, placebo-controlled study with midodrine. Am. J. Med. 1993, 95, 38–48. [Google Scholar] [CrossRef]
- Ramirez, C.E.; Okamoto, L.E.; Arnold, A.C.; Gamboa, A.; Diedrich, A.; Choi, L.; Raj, S.R.; Robertson, D.; Biaggioni, I.; Shibao, C.A. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension 2014, 64, 1235–1240. [Google Scholar] [CrossRef]
- Okamoto, L.E.; Shibao, C.A.; Gamboa, A.; Diedrich, A.; Raj, S.R.; Black, B.K.; Robertson, D.; Biaggioni, I. Synergistic Pressor Effect of Atomoxetine and Pyridostigmine in Patients with Neurogenic Orthostatic Hypotension. Hypertension 2019, 73, 235–241. [Google Scholar] [CrossRef]
- Arnold, A.C.; Raj, S.R. Orthostatic Hypotension: A Practical Approach to Investigation and Management. Can. J. Cardiol. 2017, 33, 1725–1728. [Google Scholar] [CrossRef]
- Fanciulli, A.; Goebel, G.; Metzler, B.; Sprenger, F.; Poewe, W.; Wenning, G.K.; Seppi, K. Elastic abdominal binders attenuate orthostatic hypotension in Parkinson’s disease. Mov. Disord. Clin. Pract. 2016, 3, 156–160. [Google Scholar] [CrossRef]
- Study Assessing Safety and Therapeutic Activity of AFFITOPE® PD01A and PD03A in Patients with Early MSA (AFF009). Available online: https://clinicaltrials.gov/ct2/show/NCT02270489 (accessed on 20 February 2020).
- Progression Rate of MSA Under EGCG Supplementation as Anti-Aggregation-Approach (PROMESA). Available online: https://clinicaltrials.gov/ct2/show/NCT02008721 (accessed on 20 February 2020).
- Study of NBMI Treatment in Patients with Atypical Parkinsons (PSP or MSA) (EMERA006). Available online: https://clinicaltrials.gov/ct2/show/NCT04184063 (accessed on 20 February 2020).
- Study of BHV-3241 in Subjects with Multiple System Atrophy (M-STAR). Available online: https://clinicaltrials.gov/ct2/show/NCT03952806 (accessed on 20 February 2020).
- Rituximab for Multiple System Atrphy. Available online: https://clinicaltrials.gov/ct2/show/NCT04004819 (accessed on 20 February 2020).
- Safety and Tolerability of CS10BR05 Inj. in Subjects with Multiple System Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT03265444 (accessed on 20 February 2020).
- Mesenchymal Stem Cell Therapy in Multiple System Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT02315027 (accessed on 20 February 2020).
- Neurologic Stem Cell Treatment Study (NEST). Available online: https://clinicaltrials.gov/ct2/show/NCT02795052 (accessed on 20 February 2020).
- Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of BIIB101 Administered to Adults with Multiple System Atrophy (HORIZON). Available online: https://clinicaltrials.gov/ct2/show/NCT04165486 (accessed on 20 February 2020).
- Treatment of Parkinson Disease and Multiple System Atrophy Using Intranasal Insulin. Available online: https://clinicaltrials.gov/ct2/show/NCT02064166 (accessed on 20 February 2020).
- A Futility Trial of Sirolimus in Multiple System Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT03589976 (accessed on 20 February 2020).
- Inosine 5’-Monophosphate to Raise of Serum Uric Acid Level in Patients with Multiple System Atrophy: A Multi-center, Randomized Controlled, Double Blind, Parallel Assigned Clinical Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT03403309 (accessed on 20 February 2020).
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Mandler, M.; Valera, E.; Rockenstein, E.; Mante, M.; Weninger, H.; Patrick, C.; Adame, A.; Schmidhuber, S.; Santic, R.; Schneeberger, A.; et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol. Neurodegener. 2015, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- 2 March 2018 – Affiris Announces Results Of A Phase I Clinical Study Using Affitopes® Pd01a And Pd03a, Confirming Safety And Tolerability For Both Compounds As Well As Immunogenicity For Pd01a. Available online: https://affiris.com/investors/ (accessed on 20 February 2020).
- 14 May 2018 – Affiris Announces Encouraging Long-term Data from A Series of First-in-human Studies Using Affitope® Pd01a Targeting Oligomeric Alpha-synuclein In Early Parkinson’s Disease Patients. Available online: https://affiris.com/investors/ (accessed on 20 February 2020).
- 27 January 2020 Affiris Announces Fda Response To Its Pre-ind Submission For Phase 2 Trial With Affitope® Pd01 In Early Parkinson’s Disease Patients. Available online: https://affiris.com/investors/ (accessed on 20 February 2020).
- A Study to Evaluate the Efficacy of Prasinezumab (RO7046015/PRX002) in Participants with Early Parkinson’s Disease (PASADENA). Available online: https://clinicaltrials.gov/ct2/show/NCT03100149 (accessed on 20 February 2020).
- Study of UB-312 in Healthy Participants and Parkinson’s Disease Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT04075318 (accessed on 20 February 2020).
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive Immunization Reduces Behavioral and Neuropathological Deficits in an Alpha-Synuclein Transgenic Model of Lewy Body Disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Spencer, B.; Fields, J.A.; Trinh, I.; Adame, A.; Mante, M.; Rockenstein, E.; Desplats, P.; Masliah, E. Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy. Acta Neuropathol. Commun. 2017, 5, 2. [Google Scholar] [CrossRef]
- Single Ascending Dose Study of MEDI1341 in Healthy Volunteers. Available online: https://clinicaltrials.gov/ct2/show/NCT03272165 (accessed on 20 February 2020).
- A Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of BIIB054 in Japanese Participants with Parkinson’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03716570 (accessed on 20 February 2020).
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Böddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E.; Ehrnhoefer, D.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Levites, Y.; Weinreb, O.; Maor, G.; Youdim, M.B.; Mandel, S. Green tea polyphenol (–)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem. 2001, 78, 1073–1082. [Google Scholar] [CrossRef]
- Levin, J.; Maaß, S.; Schuberth, M.; Giese, A.; Oertel, W.H.; Poewe, W.; Trenkwalder, C.; Wenning, G.K.; Mansmann, U.; Südmeyer, M.; et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2019, 18, 724–735. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef]
- Garvin, A.H.; Weckbecker, D.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b modulates α-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov. Disord. 2018, 34, 255–263. [Google Scholar] [CrossRef]
- A First-in-Human Study of Single and Multiple Doses of anle138b in Healthy Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT04208152 (accessed on 20 February 2020).
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.; Stefanova, N.; Adlard, P.; Bradbury, M.; Stamler, D. PBT434 Prevents α-synuclein Aggregation, Neuron Loss, Motor Dysfunction and Reduces Glial Cell Inclusions in a Transgenic Mouse Model of Multiple System Atrophy (P5. 8-006). Neurology 2019, 92 (Suppl. 15). [Google Scholar]
- Stamler, D.; Bradbury, M.; Wong, C.; Offman, E. A First in Human Study of PBT434, a Novel Small Molecule Inhibitor of α-Synuclein Aggregation (S4. 001). Neurology 2019, 92 (Suppl. 15). [Google Scholar]
- Zhao, H.; John, N.; Delic, V.; Ikeda-Lee, K.; Kim, A.; Weihofen, A.; Swayze, E.E.; Kordasiewicz, H.B.; West, A.B.; Volpicelli-Daley, L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol. Ther.-Nucleic Acids 2017, 8, 508–519. [Google Scholar] [CrossRef]
- A Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of BIIB094 in Adults With Parkinson’s Disease (REASON). Available online: https://clinicaltrials.gov/ct2/show/NCT03976349 (accessed on 20 February 2020).
- Vidal-Martinez, G.; Segura-Ulate, I.; Yang, B.; Diaz-Pacheco, V.; Barragan, J.A.; Esquivel, J.D.-L.; Chaparro, S.A.; Vargas-Medrano, J.; Perez, R.G. FTY720-Mitoxy reduces synucleinopathy and neuroinflammation, restores behavior and mitochondria function, and increases GDNF expression in Multiple System Atrophy mouse models. Exp. Neurol. 2020, 325, 113120. [Google Scholar] [CrossRef]
- Vargas-Medrano, J.; Krishnamachari, S.; Villanueva, E.; Godfrey, W.H.; Lou, H.; Chinnasamy, R.; Arterburn, J.B.; Perez, R.G. Novel FTY720-Based Compounds Stimulate Neurotrophin Expression and Phosphatase Activity in Dopaminergic Cells. ACS Med. Chem. Lett. 2014, 5, 782–786. [Google Scholar] [CrossRef]
- Vargas-Medrano, J.; Segura-Ulate, I.; Yang, B.; Chinnasamy, R.; Arterburn, J.B.; Perez, R.G. FTY720-Mitoxy reduces toxicity associated with MSA-like α-synuclein and oxidative stress by increasing trophic factor expression and myelin protein in OLN-93 oligodendroglia cell cultures. Neuropharmacology 2019, 158, 107701. [Google Scholar] [CrossRef]
- Vargas-Medrano, J.; Yang, B.; Garza, N.T.; Segura-Ulate, I.; Perez, R.G. Up-regulation of protective neuronal MicroRNAs by FTY720 and novel FTY720-derivatives. Neurosci. Lett. 2019, 690, 178–180. [Google Scholar] [CrossRef]
- Benskey, M.J.; Perez, R.G.; Manfredsson, F.P. The contribution of alpha synuclein to neuronal survival and function–Implications for Parkinson’s disease. J. Neurochem. 2016, 137, 331–359. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef]
- Stefanova, N.; Georgievska, B.; Eriksson, H.; Poewe, W.; Wenning, G.K. Myeloperoxidase Inhibition Ameliorates Multiple System Atrophy-Like Degeneration in a Transgenic Mouse Model. Neurotox. Res. 2011, 21, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Boireau, A.; Bordier, F.; Dubédat, P.; Pény, C.; Impérato, A. Thalidomide reduces MPTP-induced decrease in striatal dopamine levels in mice. Neurosci. Lett. 1997, 234, 123–126. [Google Scholar] [CrossRef]
- Çalışma, B.İ. The neuroprotective effects of rituximab in rat spinal cord injury model: An immunohistochemical study. Turk. Neurosurg. 2013, 23, 783–790. [Google Scholar]
- Brudek, T.; Winge, K.; Folke, J.; Christensen, S.; Fog, K.; Pakkenberg, B.; Pedersen, L. Østergaard Autoimmune antibody decline in Parkinson’s disease and Multiple System Atrophy; a step towards immunotherapeutic strategies. Mol. Neurodegener. 2017, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Bang, G.; Lee, B.R.; Kim, H.O.; Lee, P.H. Neuroprotective Effect of Human Mesenchymal Stem Cells in an Animal Model of Double Toxin-Induced Multiple System Atrophy Parkinsonism. Cell Transplant. 2011, 20, 827–836. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Panchalingam, K.M.; Rodrigues, A.J.; Mendes-Pinheiro, B.; Anjo, S.; Manadas, B.; Behie, L.A.; Sousa, N.; Salgado, A.J. Impact of the secretome of human mesenchymal stem cells on brain structure and animal behavior in a rat model of Parkinson’s disease. Stem Cells Transl. Med. 2017, 6, 634–646. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Park, H.-J.; Lee, G.; Bang, O.Y.; Ahn, Y.H.; Joe, E.; Kim, H.O.; Lee, P.H. Neuroprotective effects of human mesenchymal stem cells on dopaminergic neurons through anti-inflammatory action. Glia 2009, 57, 13–23. [Google Scholar] [CrossRef]
- Park, H.J.; Oh, S.H.; Na Kim, H.; Jung, Y.J.; Lee, P.H. Mesenchymal stem cells enhance α-synuclein clearance via M2 microglia polarization in experimental and human parkinsonian disorder. Acta Neuropathol. 2016, 132, 685–701. [Google Scholar] [CrossRef]
- Lee, P.H.; Kim, J.; Bang, O.; Ahn, Y.; Joo, I.; Huh, K. Autologous Mesenchymal Stem Cell Therapy Delays the Progression of Neurological Deficits in Patients with Multiple System Atrophy. Clin. Pharmacol. Ther. 2008, 83, 723–730. [Google Scholar] [CrossRef]
- Lee, P.H.; Lee, J.E.; Kim, H.-S.; Song, S.K.; Lee, H.S.; Nam, H.S.; Cheong, J.-W.; Jeong, Y.; Park, H.-J.; Kim, D.; et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann. Neurol. 2012, 72, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-A.; Liu, R.-S.; Lirng, J.-F.; Yang, B.-H.; Chang, C.-H.; Wang, Y.-C.; Wu, Y.-S.; Ho, J.H.-C.; Lee, O.K.; Soong, B.-W. Treatment of Spinocerebellar Ataxia with Mesenchymal Stem Cells: A Phase I/IIa Clinical Study. Cell Transplant. 2017, 26, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Eckert, M.A.; Riazifar, H.; Kang, D.-K.; Agalliu, D.; Zhao, W. From Blood to the Brain: Can Systemically Transplanted Mesenchymal Stem Cells Cross the Blood-Brain Barrier? Stem Cells Int. 2013, 2013, 1–7. [Google Scholar] [CrossRef] [PubMed]
- De Mello, N.P.; Orellana, A.M.; Mazucanti, C.H.; Lima, G.D.M.; Scavone, C.; Kawamoto, E.M. Insulin and Autophagy in Neurodegeneration. Front. Mol. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Canron, M.-H.; Vital, A.; Bezard, E.; Li, Y.; Greig, N.H.; Gulyani, S.; Kapogiannis, D.; Fernagut, P.-O.; Meissner, W.G. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain 2017, 140, 1420–1436. [Google Scholar] [CrossRef]
- Nameni, G.; Farhangi, M.A.; Hajiluian, G.; Shahabi, P.; Abbasi, M.M.; Hajilouian, G. Insulin deficiency: A possible link between obesity and cognitive function. Int. J. Dev. Neurosci. 2017, 59, 15–20. [Google Scholar] [CrossRef]
- Novak, P.; Maldonado, D.A.P.; Novak, V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS ONE 2019, 14, e0214364. [Google Scholar] [CrossRef]
- Bai, X.; Wey, M.C.-Y.; Fernandez, E.; Hart, M.J.; Gelfond, J.; Bokov, A.; Rani, S.; Strong, R. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age-related Dis. 2015, 5, 28743. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Floto, R.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef]
- Palma, J.-A.; Martinez, J.; Barnes, E.; Simon, S.; Norcliffe-Kaufmann, L.; Kaufmann, H. A Futility Trial of Sirolimus in Multiple System Atrophy: Protocol, Recruitment and Preliminary Adverse Event Profile (P3.8-019). Neurology 2019, 92 (Suppl. 15). [Google Scholar]
- Cao, B.; Wei, Q.-Q.; Ou, R.; Yang, J.; Shang, H. Association of serum uric acid level with cognitive function among patients with multiple system atrophy. J. Neurol. Sci. 2015, 359, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, D.-S.; An, C.-Y.; Liu, Y.-Z.; Liu, X.-H.; Zhang, F.; Ning, L.-N.; Li, C.-L.; Ma, C.-M.; Hu, R.-T. Association between serum uric acid level and multiple system atrophy: A meta-analysis. Clin. Neurol. Neurosurg. 2018, 169, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ng, S.Y.-E.; Chia, N.S.-Y.; Acharyya, S.; Setiawan, F.; Lu, Z.-H.; Ng, E.; Tay, K.-Y.; Au, W.-L.; Tan, E.-K. Serum uric acid level and its association with motor subtypes and non-motor symptoms in early Parkinson’s disease: PALS study. Park. Relat. Disord. 2018, 55, 50–54. [Google Scholar] [CrossRef]
- Glat, M.J.; Stefanova, N.; Wenning, G.K.; Offen, D. Genes to treat excitotoxicity ameliorate the symptoms of the disease in mice models of multiple system atrophy. J. Neural Transm. 2020, 127, 205–212. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; Mccord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Bessa, M.; Plaitakis, A. Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J. Neurosci. Res. 2005, 79, 65–73. [Google Scholar] [CrossRef]
- Guo, H.; Lai, L.; Butchbach, M.E.R.; Stockinger, M.P.; Shan, X.; Bishop, G.A.; Lin, C.-L.G. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 2003, 12, 2519–2532. [Google Scholar] [CrossRef]
- Barca, E.; Kleiner, G.; Tang, G.; Ziosi, M.; Tadesse, S.; Masliah, E.; Louis, E.D.; Faust, P.; Kang, U.J.; Torres, J.; et al. Decreased Coenzyme Q10 Levels in Multiple System Atrophy Cerebellum. J. Neuropathol. Exp. Neurol. 2016, 75, 663–672. [Google Scholar] [CrossRef]
- Nakamoto, F.K.; Okamoto, S.; Mitsui, J.; Sone, T.; Ishikawa, M.; Yamamoto, Y.; Kanegae, Y.; Nakatake, Y.; Imaizumi, K.; Ishiura, H.; et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Koguchi, K.; Momose, T.; Takahashi, M.; Matsukawa, T.; Yasuda, T.; Tokushige, S.-I.; Ishiura, H.; Goto, J.; Nakazaki, S.; et al. Three-Year Follow-Up of High-Dose Ubiquinol Supplementation in a Case of Familial Multiple System Atrophy with Compound Heterozygous COQ2 Mutations. Cerebellum 2017, 16, 664–672. [Google Scholar] [CrossRef] [PubMed]
- UMIN.ac.jp. Available online: https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000019376 (accessed on 20 February 2020).
- Mei, F.; Fancy, S.P.J.; Shen, Y.-A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef]
- Göttle, P.; Förster, M.; Weyers, V.; Küry, P.; Rejdak, K.; Hartung, H.-P.; Kremer, D. An unmet clinical need: Roads to remyelination in MS. Neurol. Res. Pract. 2019, 1, 21. [Google Scholar] [CrossRef]
- Kremer, D.; Göttle, P.; Flores-Rivera, J.; Hartung, H.-P.; Küry, P. Remyelination in multiple sclerosis: From concept to clinical trials. Curr. Opin. Neurol. 2019, 32, 378–384. [Google Scholar] [CrossRef]

{kind=link}
| Symptoms | Drug Therapy | Clinical Trials | ||
|---|---|---|---|---|
| Phase | NCT Number | Ref. | ||
| Parkinsonism | MAO-B inhibitor safinamide | 2 | NCT03753763 | [50] |
| Cerebellar ataxia | NMDA receptor modulator Tllsh2910 | 3 | NCT03901638 | [51] |
| Orthostatic hypotension | α1-receptor inhibitor midodrine | 1 | NCT02897063 | [52] |
| Norepinephrine prodrug droxidopa (l-threo DOPS) | 1 | NCT02897063 | [52] | |
| 4 | NCT02586623 | [53] | ||
| Selective NET inhibitor atomexetine | 2 | NCT02796209 | [54] | |
| NRI ampreloxetine (TD-9855) | 2 | NCT03750552 | [55] | |
| Therapeutic Targets | Drug Therapy | Clinical Trials | ||
|---|---|---|---|---|
| Phase | NCT Number | Ref. | ||
| α-Synuclein aggregation | Vaccines Affitope PD01A and PD03A | 1 | NCT02270489 | [77] |
| Epigallocatechin gallate (EGCG) | 3 | NCT02008721 | [78] | |
| Chelator and antioxidant NMBI (Irminix) | 2 | NCT04184063 | [79] | |
| Neuroinflammation | MPO inhibitor verdiperstat | 3 | NCT03952806 | [80] |
| CD20-antibody rituximab | 2 | NCT04004819 | [81] | |
| Neuronal loss | Bone marrow-derived mesenchymal stem cells (MSCs) | 1 | NCT03265444 | [82] |
| 1 | NCT02315027 | [83] | ||
| n.a. | NCT02795052 | [84] | ||
| 1 | NCT04165486 | [85] | ||
| Intranasal insulin | 2 | NCT02064166 | [86] | |
| mTOR inhibitor sirolimus | 2 | NCT03589976 | [87] | |
| Antioxidant inosine 5′-monophosphate | 2 | NCT03403309 | [88] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mészáros, L.; Hoffmann, A.; Wihan, J.; Winkler, J. Current Symptomatic and Disease-Modifying Treatments in Multiple System Atrophy. Int. J. Mol. Sci. 2020, 21, 2775. https://doi.org/10.3390/ijms21082775
Mészáros L, Hoffmann A, Wihan J, Winkler J. Current Symptomatic and Disease-Modifying Treatments in Multiple System Atrophy. International Journal of Molecular Sciences. 2020; 21(8):2775. https://doi.org/10.3390/ijms21082775
Chicago/Turabian StyleMészáros, Lisa, Alana Hoffmann, Jeanette Wihan, and Jürgen Winkler. 2020. "Current Symptomatic and Disease-Modifying Treatments in Multiple System Atrophy" International Journal of Molecular Sciences 21, no. 8: 2775. https://doi.org/10.3390/ijms21082775
APA StyleMészáros, L., Hoffmann, A., Wihan, J., & Winkler, J. (2020). Current Symptomatic and Disease-Modifying Treatments in Multiple System Atrophy. International Journal of Molecular Sciences, 21(8), 2775. https://doi.org/10.3390/ijms21082775