Fluorinated Molecules and Nanotechnology: Future ‘Avengers’ against the Alzheimer’s Disease?




Abstract

1. Introduction
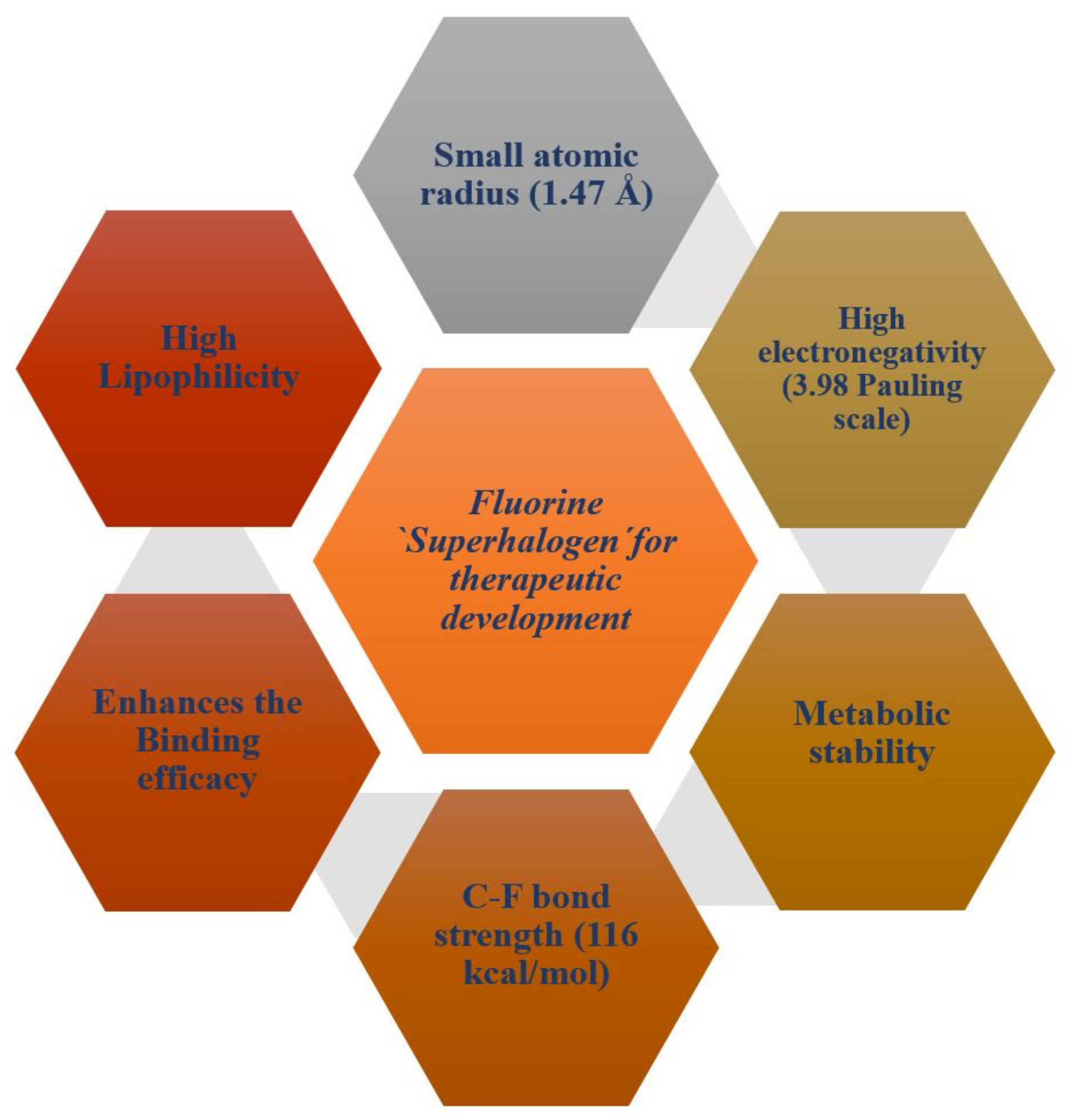
2. The Role of Fluorine in the Development of Therapeutic Drugs
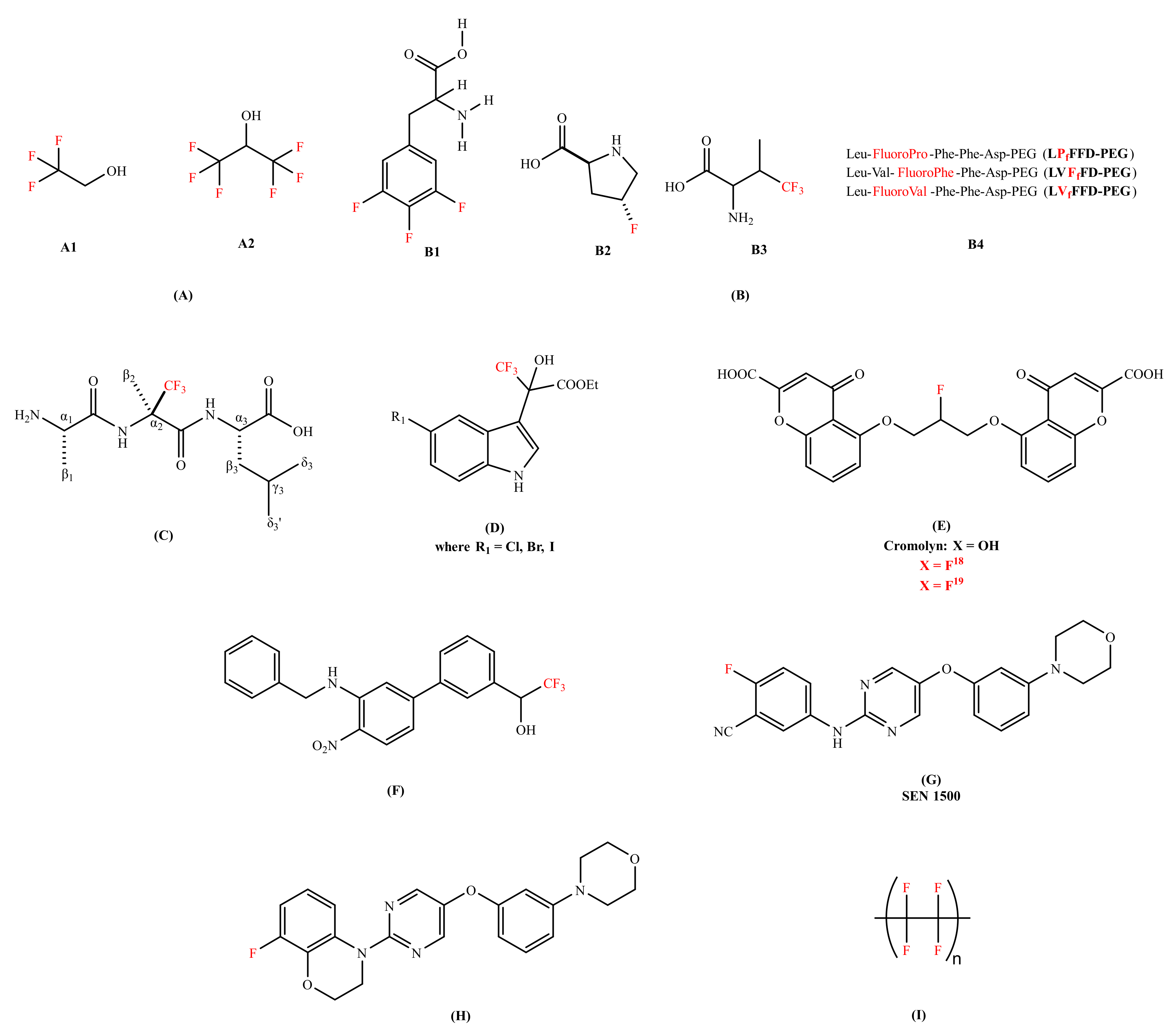
2.1. The Role of Fluorine-Containing Compounds in the Modulation of Amyloid-Beta Peptide
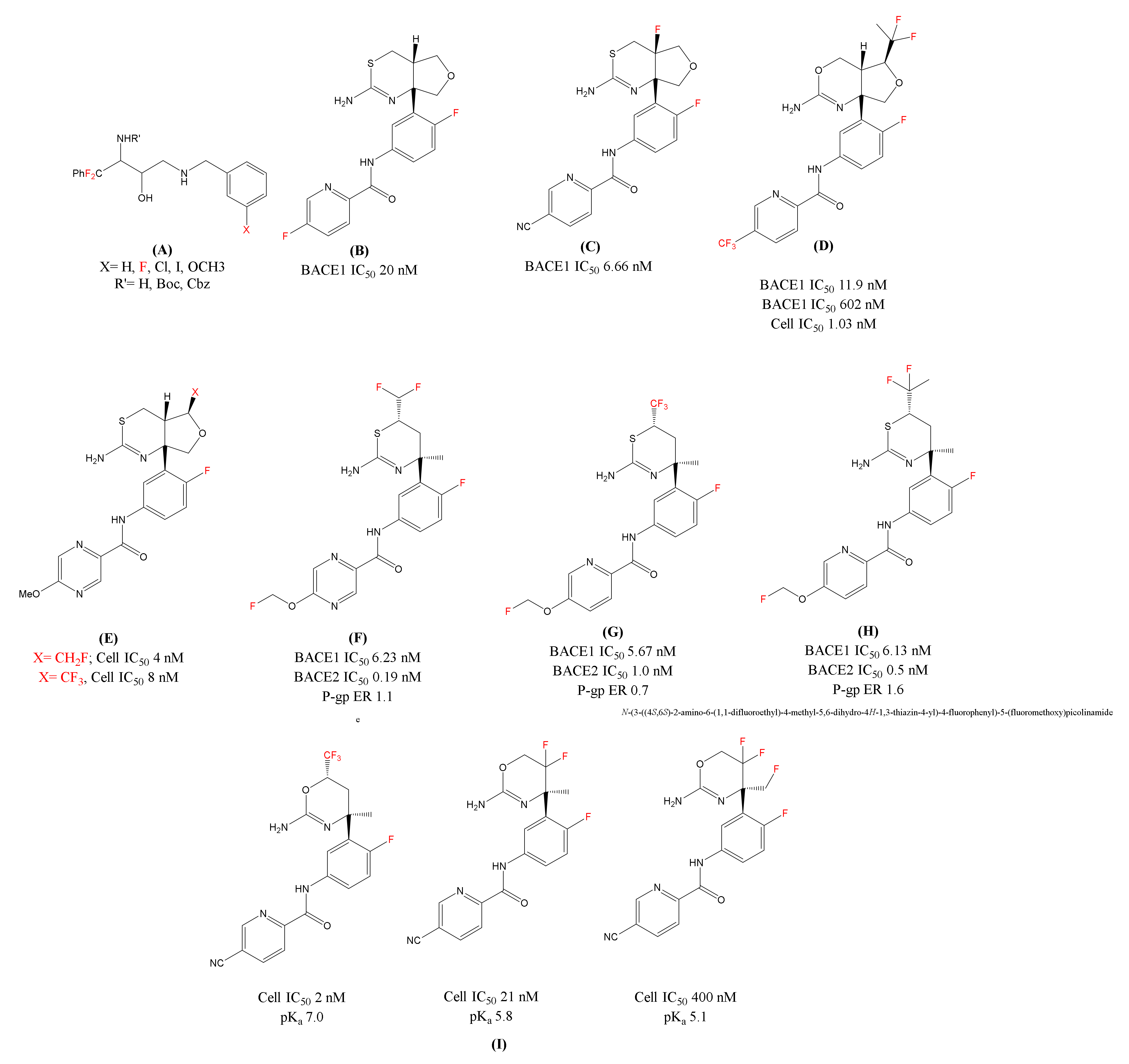
2.2. The Role of Fluorine-Containing Compounds in the Modulation of the Secretases
3. Nanoparticles Designed with Fluorine Molecules for the Treatment of Alzheimer’s Disease
4. Conclusion and Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α | Alpha |
| β | Beta |
| Aβ | Amyloid-beta |
| AD | Alzheimer’s disease |
| AFM | Atomic force microscopy |
| BACE | β-Site amyloid precursor protein cleaving enzyme |
| BBB | Blood-brain barrier |
| CD | Circular dichroism |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DFT | Density functional theory |
| EtOH | Ethanol |
| FAD | Familial Alzheimer’s disease |
| FTIR | Fourier-transform infrared spectroscopy |
| HFIP | Hexafluoroisopropanl |
| LTP | Long-term potentiation |
| NBO | Natural bond orbital |
| NMR | Nuclear magnetic resonance |
| NPs | Nanoparticles |
| Teflon | Polytetrafluoroethylene |
| TEM | Transmission electron microscopy |
| TFE | Trifluoroethanol |
| Tfm | Trifluoromethylated |
| THF | Tetrahydrofuran |
| ThT | Thioflavin T |
References
- Association, A. 2015 Alzheimer’s disease facts and figures. Alzheimer’s & Dement. 2015, 11, 332–384. [Google Scholar]
- Bertram, L.; Tanzi, R.E. The genetics of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 107, 79–100. [Google Scholar] [PubMed]
- Association, A. 2019 Alzheimer’s disease facts and figures. Alzheimer’s & Dement. 2019, 15, 321–387. [Google Scholar]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Sumner Makin, O.; Serpell, L.C. Structural Characterisation of Islet Amyloid Polypeptide Fibrils. J. Mol. Biol. 2004, 335, 1279–1288. [Google Scholar] [CrossRef]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef]
- Nag, S.; Sarkar, B.; Bandyopadhyay, A.; Sahoo, B.; Sreenivasan, V.K.; Kombrabail, M.; Muralidharan, C.; Maiti, S. Nature of the amyloid-beta monomer and the monomer-oligomer equilibrium. J. Biol. Chem. 2011, 286, 13827–13833. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet (London, England) 1976, 2, 1403. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain A J. Neurol. 1976, 99, 459–496. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. & Ther. 2014, 6, 37. [Google Scholar]
- Balson, R.; Gibson, P.R.; Ames, D.; Bhathal, P.S. Tacrine-Induced Hepatotoxicity. CNS Drugs 1995, 4, 168–181. [Google Scholar] [CrossRef]
- Maia, M.A.; Sousa, E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals (Basel) 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.D.C. Natural Compounds for Alzheimer’s Disease Therapy: A Systematic Review of Preclinical and Clinical Studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef]
- Gladysz, J.A.; Curran, D.P. Fluorous chemistry: From biphasic catalysis to a parallel chemical universe and beyond. Tetrahedron 2002, 58, 3823–3825. [Google Scholar] [CrossRef]
- Yi, W.-B.; Ma, J.-J.; Jiang, L.-Q.; Cai, C.; Zhang, W. Synthesis and uses of fluorous and highly fluorinated macrocyclic and spherical molecules. J. Fluor. Chem. 2014, 157, 84–105. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Potschka, H. Targeting the brain--surmounting or bypassing the blood-brain barrier. Handb. Exp. Pharmacol. 2010, 197, 411–431. [Google Scholar]
- Loureiro, J.A.; Gomes, B.; Coelho, M.A.; Do Carmo Pereira, M.; Rocha, S. Targeting nanoparticles across the blood-brain barrier with monoclonal antibodies. Nanomedicine (London) 2014, 9, 709–722. [Google Scholar] [CrossRef]
- Joshi, S.A.; Chavhan, S.S.; Sawant, K.K. Rivastigmine-loaded PLGA and PBCA nanoparticles: Preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur. J. Pharm. Biopharm. 2010, 76, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, E.; Ettcheto, M.; Egea, M.A.; Espina, M.; Cano, A.; Calpena, A.C.; Camins, A.; Carmona, N.; Silva, A.M.; Souto, E.B.; et al. Memantine loaded PLGA PEGylated nanoparticles for Alzheimer’s disease: In vitro and in vivo characterization. J. Nanobiotechnology 2018, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.S.; Paramakrishnan, N.; Suresh, B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008, 1200, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.; Le Droumaguet, B.; Nicolas, J.; Hashemi, S.H.; Wu, L.P.; Moghimi, S.M.; Couvreur, P.; Andrieux, K. Nanotechnologies for Alzheimer’s disease: Diagnosis, therapy, and safety issues. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 521–540. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Y.; Yang, Y.; Sun, L.; Han, D.; Li, H.; Wang, C. Pharmacological and toxicological target organelles and safe use of single-walled carbon nanotubes as drug carriers in treating Alzheimer disease. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 427–441. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef]
- Arya, M.A.; Kumar, M.; Sabitha, M.; Menon, K.; Nair, S. Nanotechnology approaches for enhanced CNS delivery in treating Alzheimer’s disease. J. Drug Deliv. Sci. Technol. 2019, 51, 297–309. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Gomes, B.; Fricker, G.; Coelho, M.A.N.; Rocha, S.; Pereira, M.C. Cellular uptake of PLGA nanoparticles targeted with anti-amyloid and anti-transferrin receptor antibodies for Alzheimer’s disease treatment. Colloids Surf. B Biointerfaces 2016, 145, 8–13. [Google Scholar] [CrossRef]
- Karthivashan, G.; Ganesan, P.; Park, S.Y.; Kim, J.S.; Choi, D.K. Therapeutic strategies and nano-drug delivery applications in management of ageing Alzheimer’s disease. Drug Deliv. 2018, 25, 307–320. [Google Scholar] [CrossRef]
- Rocha, S.; Thunemann, A.F.; Pereira Mdo, C.; Coelho, M.; Mohwald, H.; Brezesinski, G. Influence of fluorinated and hydrogenated nanoparticles on the structure and fibrillogenesis of amyloid beta-peptide. Biophys. Chem. 2008, 137, 35–42. [Google Scholar] [CrossRef]
- Saraiva , A.M.; Cardoso , I.; Pereira, M.C.; Coelho, M.A.N.; Saraiva, M.J.; Möhwald, H.; Brezesinski, G. Controlling Amyloid-β Peptide(1–42) Oligomerization and Toxicity by Fluorinated Nanoparticles. ChemBioChem 2010, 11, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Loureiro, J.A.; Coelho, M.A.; Pereira Mdo, C. The Potential Effect of Fluorinated Compounds in the Treatment of Alzheimer’s Disease. Curr. Pharm. Des. 2015, 21, 5725–5735. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem.—A Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L.; Filler, R. Recent Advances in the Biomedicinal Chemistry of Fluorine-Containing Compounds. In Biomedical Frontiers of Fluorine Chemistry; American Chemical Society: Washington, DC, USA, 1996; Volume 639, pp. 1–24. [Google Scholar]
- O’Hagan, D.S.; Rzepa, H. Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997, 7, 645–652. [Google Scholar] [CrossRef]
- Zhang, X.J.; Lai, T.B.; Kong, R.Y. Biology of fluoro-organic compounds. Top. Curr. Chem. 2012, 308, 365–404. [Google Scholar]
- Resnati, G. Aspects of the medicinal chemistry of fluoroorganic compounds. Part, I. Farm. (Soc. Chim. Ital. 1989) 1990, 45, 1043–1066. [Google Scholar]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; Del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Bégué, J.-P.; Bonnet-Delpon, D. Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural products. J. Fluor. Chem. 2006, 127, 992–1012. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medical chemistry. J. Enzime Inhib. Med Chem. 2005, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Haranahalli, K.; Honda, T.; Ojima, I. Recent progress in the strategic incorporation of fluorine into medicinally active compounds. J. Fluor. Chem. 2019, 217, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science (New York) 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Shengguo, S.; Adeboye, A. Fluorinated Molecules as Drugs and Imaging Agents in the CNS. Curr. Top. Med. Chem. 2006, 6, 1457–1464. [Google Scholar]
- Pan, Y. The Dark Side of Fluorine. ACS Medicinal Chem. Lett. 2019, 10, 1016–1019. [Google Scholar] [CrossRef]
- Wermers, R.A.; Cooper, K.; Razonable, R.R.; Deziel, P.J.; Whitford, G.M.; Kremers, W.K.; Moyer, T.P. Fluoride excess and periostitis in transplant patients receiving long-term voriconazole therapy. Clin. Infect. Dis. 2011, 52, 604–611. [Google Scholar] [CrossRef]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M. Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 2002, 4, 155–174. [Google Scholar] [CrossRef] [PubMed]
- Carrotta, R.; Manno, M.; Giordano, F.M.; Longo, A.; Portale, G.; Martorana, V.; Biagio, P.L.S. Protein stability modulated by a conformational effector: Effects of trifluoroethanol on bovine serum albumin. Phys. Chem. Chem. Phys. 2009, 11, 4007–4018. [Google Scholar] [CrossRef]
- Vieira, E.P.; Hermel, H.; Möhwald, H. Change and stabilization of the amyloid-β(1–40) secondary structure by fluorocompounds. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2003, 1645, 6–14. [Google Scholar] [CrossRef]
- Asante, V.; Mortier, J.; Wolber, G.; Koksch, B. Impact of fluorination on proteolytic stability of peptides: A case study with alpha-chymotrypsin and pepsin. Amino Acids 2014, 46, 2733–2744. [Google Scholar] [CrossRef] [PubMed]
- Koksch, B.; Sewald, N.; Hofmann, H.J.; Burger, K.; Jakubke, H.D. Proteolytically stable peptides by incorporation of alpha-Tfm amino acids. J. Pept. Sci. An Off. Publ. Eur. Pept. Soc. 1997, 3, 157–167. [Google Scholar]
- Salwiczek, M.; Nyakatura, E.K.; Gerling, U.I.; Ye, S.; Koksch, B. Fluorinated amino acids: Compatibility with native protein structures and effects on protein-protein interactions. Chem. Soc. Rev. 2012, 41, 2135–2171. [Google Scholar] [CrossRef] [PubMed]
- Feytens, D.; Chaume, G.; Chassaing, G.; Lavielle, S.; Brigaud, T.; Byun, B.J.; Kang, Y.K.; Miclet, E. Local control of the cis-trans isomerization and backbone dihedral angles in peptides using trifluoromethylated pseudoprolines. J. Phys. Chem. B 2012, 116, 4069–4079. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Crespo, R.; Börner, H.; Martins, P.M.; Rocha, F.A.; Coelho, M.; Pereira, M.C.; Rocha, S. Fluorinated beta-sheet breaker peptides. J. Mater. Chem. B 2014, 2, 2259–2264. [Google Scholar] [CrossRef]
- Botz, A.; Gasparik, V.; Devillers, E.; Hoffmann, A.R.; Caillon, L.; Chelain, E.; Lequin, O.; Brigaud, T.; Khemtemourian, L. (R)-α-trifluoromethylalanine containing short peptide in the inhibition of amyloid peptide fibrillation. Biopolymers 2015, 104, 601–610. [Google Scholar] [CrossRef]
- Török, M.; Abid, M.; Mhadgut, S.C.; Török, B. Organofluorine Inhibitors of Amyloid Fibrillogenesis. Biochemistry 2006, 45, 5377–5383. [Google Scholar] [CrossRef]
- Sood, A.; Abid, M.; Hailemichael, S.; Foster, M.; Török, B.; Török, M. Effect of chirality of small molecule organofluorine inhibitors of amyloid self-assembly on inhibitor potency. Bioorganic Med. Chem. Lett. 2009, 19, 6931–6934. [Google Scholar]
- Sood, A.; Abid, M.; Sauer, C.; Hailemichael, S.; Foster, M.; Török, B.; Török, M. Disassembly of preformed amyloid beta fibrils by small organofluorine molecules. Bioorganic Med. Chem. Lett. 2011, 21, 2044–2047. [Google Scholar]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer β-fibrillogenesis by melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef]
- Elmaleh, D.R.; Shoup, T. Cromolyn Derivatives and Related Methods of Imaging and Treatment. WO 2010088455A2, 29 January 2010. [Google Scholar]
- Reed, M.A.; Yadav, A.; Banfield, S.C.; Barden, C.J. Anti-amyloid Compounds and Methods. WO 2012119035A1, 9 July 2012. [Google Scholar]
- Horwell, D.; Scopes, D. Compounds. WO 2011144577A1, 10 May 2011. [Google Scholar]
- Horwell, D.C.; Scopes, D.I.C. Compounds. WO 2011144578A1, 10 May 2011. [Google Scholar]
- Giacomelli, C.E.; Norde, W. Influence of Hydrophobic Teflon Particles on the Structure of Amyloid β-Peptide. Biomacromolecules 2003, 4, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Bela, T.; Sujaya, D.; Marianna, T. Chemistry of Small Molecule Inhibitors in Self-Assembly of Alzheimers Disease Related Amyloid-Beta Peptide. Curr. Bioact. Compd. 2008, 4, 159–174. [Google Scholar]
- Khosravan, A.; Marani, S.; Sadeghi Googheri, M.S. The effects of fluorine substitution on the chemical properties and inhibitory capacity of Donepezil anti-Alzheimer drug; density functional theory and molecular docking calculations. J. Mol. Graph. Model. 2017, 71, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.-C.; Rombouts, F.; Gijsen, H.J.M. New evolutions in the BACE1 inhibitor field from 2014 to 2018. Bioorganic & Med. Chem. Lett. 2019, 29, 761–777. [Google Scholar]
- Roberds, S.L.; Anderson, J.; Basi, G.; Bienkowski, M.J.; Branstetter, D.G.; Chen, K.S.; Freedman, S.B.; Frigon, N.L.; Games, D.; Hu, K.; et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: Implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 2001, 10, 1317–1324. [Google Scholar] [CrossRef]
- Maillard, M.C.; Hom, R.K.; Benson, T.E.; Moon, J.B.; Mamo, S.; Bienkowski, M.; Tomasselli, A.G.; Woods, D.D.; Prince, D.B.; Paddock, D.J.; et al. Design, Synthesis, and Crystal Structure of Hydroxyethyl Secondary Amine-Based Peptidomimetic Inhibitors of Human β-Secretase. J. Med. Chem. 2007, 50, 776–781. [Google Scholar] [CrossRef]
- Fustero, S.; Cuñat, A.C.; Flores, S.; Báez, C.; Oliver, J.; Cynamon, M.; Gütschow, M.; Mertens, M.D.; Delgado, O.; Tresadern, G.; et al. Design, Synthesis, and Biological Evaluation of Novel Fluorinated Ethanolamines. Chem.—A Eur. J. 2011, 17, 14772–14784. [Google Scholar] [CrossRef]
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The Potent BACE1 Inhibitor LY2886721 Elicits Robust Central Aβ Pharmacodynamic Responses in Mice, Dogs, and Humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef]
- Green, S.J.; Hembre, E.J.; Mergott, D.J.; Shi, Y.; Watson, B.M.; Winneroski Jr., L.L. BACE Inhibitors. WO 2014204730, 11 June 2014. [Google Scholar]
- Michael, D.; Simon, R.; Richards, J.; Sanderson, A.J. Tetrahydrofurane-Fused Aminohydrothiazine Derivatives which are Useful in the Treatment of Alzheimer’s Disease. WO 2016176118A1, 22 April 2016. [Google Scholar]
- Coates, D.A.; Hembre, E.J. -[3-[2-amino-5-(1,1-difluoroethyl) -4,4a,5,7-tetrahydrofuro [3,4-d][1,3]oxazin-7a-yl]-4-fluoro-phenyl]-5-(trifluoromethyl)pyridine-2-carboxamide and its (4ar,5s,7as) isomer as a selective bace1 inhibitor for treating e.g., alzheimer’s disease. WO 2017200863A1, 15 May 2017. [Google Scholar]
- Mergott, D.J.; Willis, B.A. Combination therapy of BACE-1 inhibitor and anti-N3PGLU abeta antibody. WO 2018034977, 22 February 2018. [Google Scholar]
- Dimopoulos, P.; Hall, A.; Kita, Y.; Madin, A.; Shuker, N.L. Fused aminodihydrothiaziine derivatives. WO /2012/093148, 22 February 2012. [Google Scholar]
- Hall, A.; Farthing, C.N.; Castro, P.; Fused, J. Used aminodihydrothiazine derivatives useful as bace inhibitors. WO /2012/098213, 21 January 2012. [Google Scholar]
- Kusakabe, K.; Tadano, G.; Komano, K.; Fuchino, K.; Nakahara, K. Dihydrothiazine and dihydrooxazine derivatives having BACE1 inhibitory activity. WO /2015/156421, 15 October 2015. [Google Scholar]
- Woltering, T.J.; Wostl, W.; Hilpert, H.; Rogers-Evans, M.; Pinard, E.; Mayweg, A.; Göbel, M.; Banner, D.W.; Benz, J.; Travagli, M.; et al. BACE1 inhibitors: A head group scan on a series of amides. Bioorganic Med. Chem. Lett. 2013, 23, 4239–4243. [Google Scholar]
- Lueoend, R.M.; Machauer, R.; Rueeger, H.; Veenstra, S.J. 2 -amino-4 -(pyridin-2-yl)-5, 6-dihidro-4H-1, 3-oxazine derivatives and their use as BACE-1 and/or BACE—2 inhibitors. WO /2013/027188, 28 February 2013. [Google Scholar]
- Low, J.D.; Bartberger, M.D.; Cheng, Y.; Whittington, D.; Xue, Q.; Wood, S.; Allen, J.R.; Minatti, A.E. Diastereoselective synthesis of fused cyclopropyl-3-amino-2,4-oxazine beta-amyloid cleaving enzyme (BACE) inhibitors and their biological evaluation. Bioorg. Med. Chem. Lett. 2018, 28, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Odagiri, T.; Inagaki, H.; Nagamochi, M.; Kitamura, T.; Komoriya, S.; Takahashi, H. Design, Synthesis, and Biological Evaluation of Novel 7-[(3aS,7aS)-3a-Aminohexahydropyrano [3,4-c]pyrrol-2(3H)-yl]-8-methoxyquinolines with Potent Antibacterial Activity against Respiratory Pathogens. J. Med. Chem. 2018, 61, 7234–7244. [Google Scholar] [CrossRef] [PubMed]
- Hilpert, H.; Guba, W.; Woltering, T.J.; Wostl, W.; Pinard, E.; Mauser, H.; Mayweg, A.V.; Rogers-Evans, M.; Humm, R.; Krummenacher, D.; et al. β-Secretase (BACE1) Inhibitors with High in Vivo Efficacy Suitable for Clinical Evaluation in Alzheimer’s Disease. J. Med. Chem. 2013, 56, 3980–3995. [Google Scholar] [CrossRef]
- Liang, S.H.; Southon, A.G.; Fraser, B.H.; Krause-Heuer, A.M.; Zhang, B.; Shoup, T.M.; Lewis, R.; Volitakis, I.; Han, Y.; Greguric, I.; et al. Novel Fluorinated 8-Hydroxyquinoline Based Metal Ionophores for Exploring the Metal Hypothesis of Alzheimer’s Disease. ACS Med. Chem. Lett. 2015, 6, 1025–1029. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Site of Action | Observed Effects | Models Used | Reference |
|---|---|---|---|---|
| Fluorinated alcohols (TFE and HFIP) | Aβ40 peptide | Induce conformational transition from β-sheet to α-helical structure | Stock solution of Aβ40 peptide | [51] |
| LVfFFD-PEG and LVFfFD-PEG | Aβ42 peptide | Delay the formation of the Aβ aggregates | Stock solution of Aβ42 peptide | [56] |
| (R)-α-trifluoromethylalanine | Delays the conformational transition for at least 96 h; Slows the kinetic depletion rate of Aβ monomers | Stock solution of Aβ42 peptide | [57] | |
| 5′-halogen substituted 3,3,3-trifluoromethyl-2-hydroxyl-(indol-3-yl)-propionic acid esters | Aβ40 peptide | Inhibits the fibrillogenesis; Disassemble preformed fibrils | Stock solution of Aβ40 peptide | [58,59,60] |
| Cromolyn-based fluorinated derivative | Aβ40 and Aβ42 aggregates | Inhibits the Aβ oligomerization; Displays significant brain uptake and clearance activity | Stock solution of Aβ40 and Aβ42 peptideand WT mice | [62] |
| Organofluorine inhibitor | Aβ40 peptide | Improve the cognitive impairment; Enhance the BBB permeability | Stock solution of Aβ40 peptide and mice | [63] |
| SEN 1500 | Aβ42 peptide | Demonstrates anti-aggregating capability; Blocks the toxic effect in LTP | Stock solution of Aβ42 peptide, SH-SY5Y cells and 7PA2 CM cells | [65] |
| 8-fluoro-3,4-dihydro-2H benzo [1,4] oxazine inhibitor | Inhibits Aβ aggregation; Shows excellent neuroprotective profile | Stock solution of Aβ42 peptide, SH-SY5Y cells, hippocampal slices of male young rat (6–8 weeks old) | [65] | |
| Fluorinated surface (Teflon) | Aβ40 peptide | Promotes α-helix reformation | Stock solution of Aβ40 peptide | [66] |
| Compounds | Site of Action | Observed Effects | Model Used | Reference |
|---|---|---|---|---|
| Fluorinated ethanolamines | β-secretase (BACE1) | Inhibits BACE1 activity | Enzymatic assay (human BACE1), human neuroblastoma SKNBE2 cells | [72] |
| LY-2886721 | Decreases the Aβ levels in CSF | Human. Terminated after phase 2 due to liver toxicity | [75] | |
| Fluorinated LY-2886721 | Reduces the amyloid levels | HEK293 cells (Human BACE1)PDAPP young mice | [76] | |
| 1,3 oxazine-based BACE1 inhibitor (difluoroethyl substituted analogue) | Display good BACE1/2 selectivity; Reduce Aβ levels in CSF | HEK293 cells (Both human BACE1 and BACE2) male beagle dogs | [78] | |
| Eisai’s BACE1 inhibitor [1,3] thiazine series Fluoro(methyl) analogues | Enhance the basicity and show selectivity over BACE2 | Human/Rat Aβ42; neuronal cultures of rat’s fetus brain | [80,81] | |
| Organofluorine substituted BACE1 inhibitors | Improve the drug efficacy (non-P-gp substrates) | Neuroblastoma SH-SY5Y cells, human liver microsomes, ICR mice (7–9 weeks old) | [82] | |
| Fluorinated oxazines analogues | Enhance potency and basicity; Reduce the Aβ levels at low doses | Enzymatic assays (BACE1 and BACE2), HEK293 cells, LLC-PK1 cells, female WT-mice | [88] |
| Compounds | Site of Action | Observed Effects | Model Used | Reference |
|---|---|---|---|---|
| Fluorinated nanoparticles | Aβ40 | Inhibition of amyloid fibril formation | Stock solution of Aβ40 peptide (in vitro) | [30] |
| Aβ42 | Anti-oligomeric and anti-apoptotic activity | Stock solution of Aβ42 peptide (in vitro) and SH-SY5Y cells (in vitro) | [31] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabur, M.; Loureiro, J.A.; Pereira, M.C. Fluorinated Molecules and Nanotechnology: Future ‘Avengers’ against the Alzheimer’s Disease? Int. J. Mol. Sci. 2020, 21, 2989. https://doi.org/10.3390/ijms21082989
Dabur M, Loureiro JA, Pereira MC. Fluorinated Molecules and Nanotechnology: Future ‘Avengers’ against the Alzheimer’s Disease? International Journal of Molecular Sciences. 2020; 21(8):2989. https://doi.org/10.3390/ijms21082989
Chicago/Turabian StyleDabur, Meghna, Joana A. Loureiro, and Maria Carmo Pereira. 2020. "Fluorinated Molecules and Nanotechnology: Future ‘Avengers’ against the Alzheimer’s Disease?" International Journal of Molecular Sciences 21, no. 8: 2989. https://doi.org/10.3390/ijms21082989
APA StyleDabur, M., Loureiro, J. A., & Pereira, M. C. (2020). Fluorinated Molecules and Nanotechnology: Future ‘Avengers’ against the Alzheimer’s Disease? International Journal of Molecular Sciences, 21(8), 2989. https://doi.org/10.3390/ijms21082989