Structural Characterization of Carbonic Anhydrase VIII and Effects of Missense Single Nucleotide Variations to Protein Structure and Function



Abstract
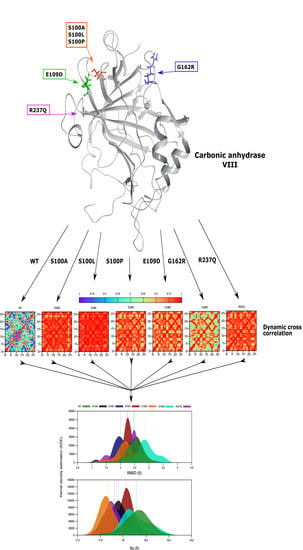
1. Introduction
2. Results and Discussion
2.1. Data Retrieval Identifies SNVs Pathogenic to CA-VIII
2.2. Functional Analysis Reveals Key Protein–Protein Interaction Residues
2.3. Sequence Analysis Identifies Residues Essential to CA-VIII Structure
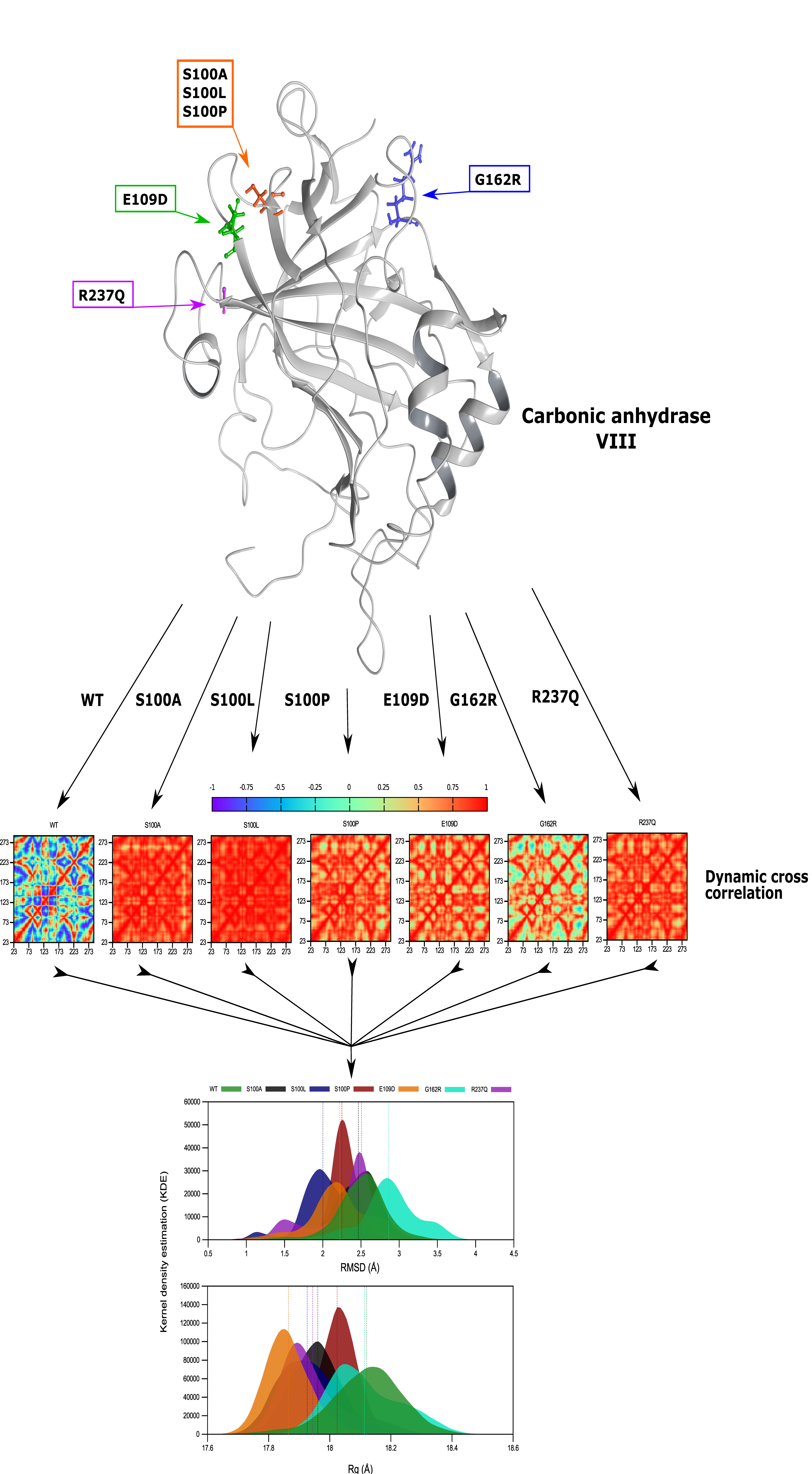
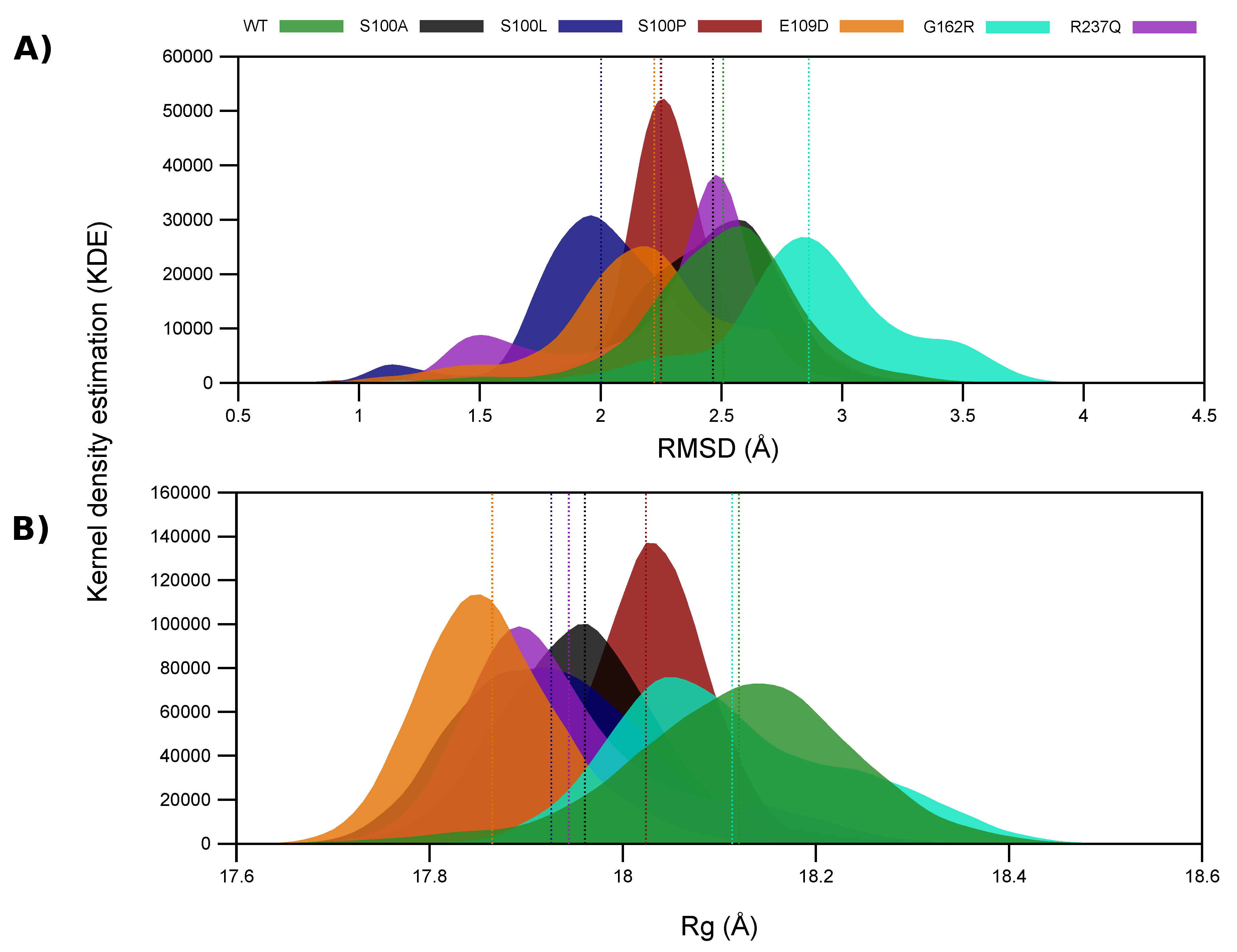
2.4. Variant Presence Causes Conformational Changes to CA-VIII
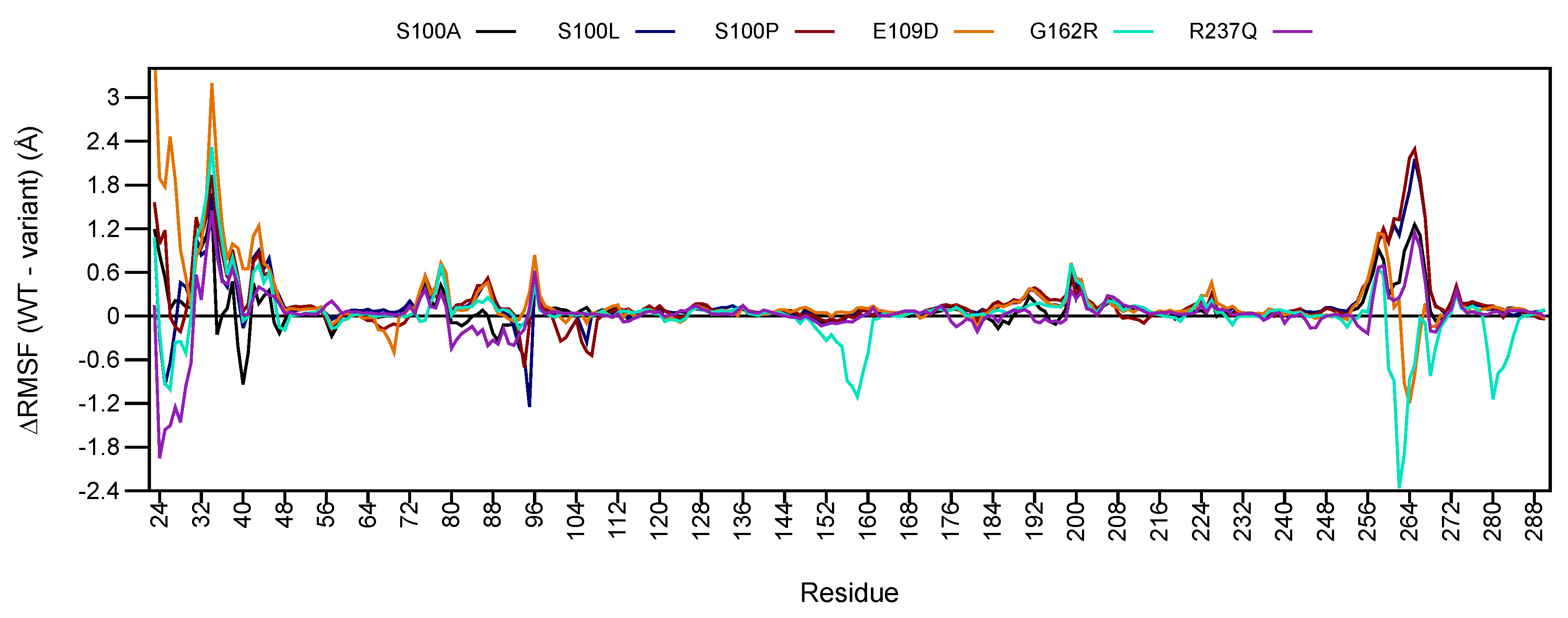
2.5. WT and Variant Proteins Show Differences to Residue Movement
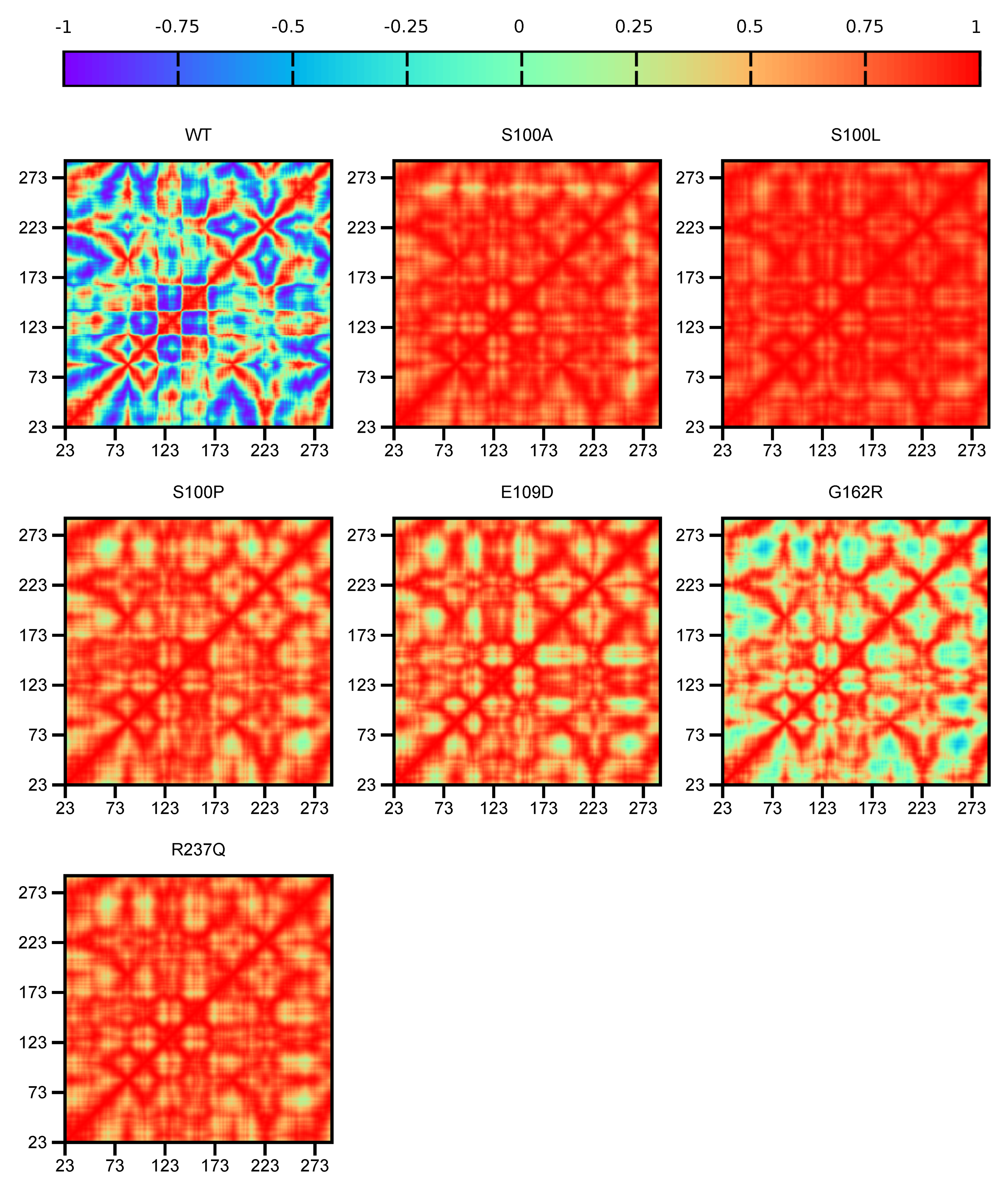
2.5.1. DCC Analysis
2.5.2. RMSF Analysis
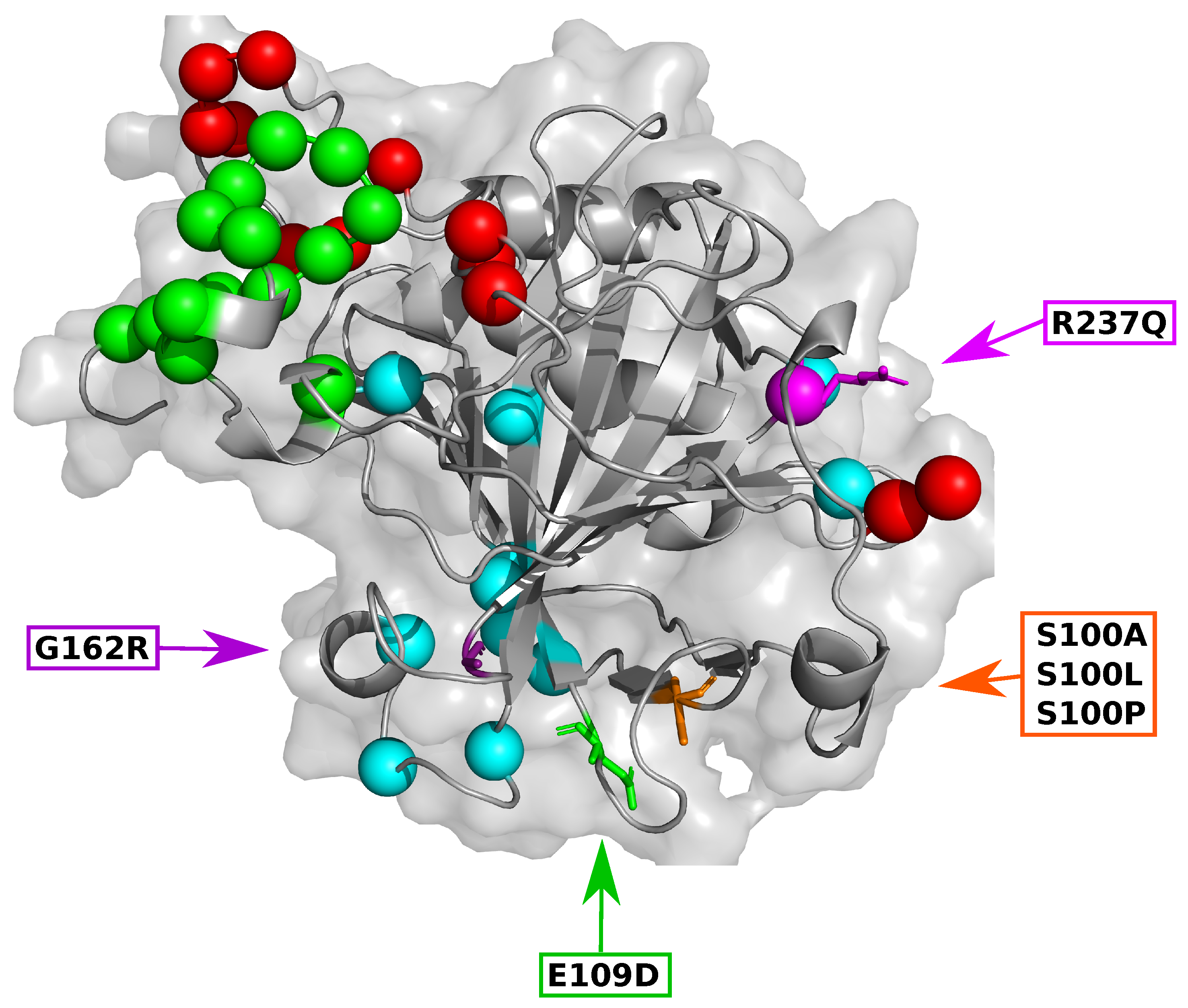
2.6. Variant Presence Is Associated with Changes to Residue Accessibility and Communication
3. Materials and Methods
3.1. Software
3.2. Homology Modelling
3.2.1. Wild-Type
3.2.2. Variants
3.3. Essential Residue Identification
3.4. Identification of CA-VIII Binding Site Residues
3.5. Molecular Dynamics
3.6. Molecular Dynamics Trajectory Analysis
Dynamic Cross Correlation (DCC)
3.7. Dynamic Residue Network Analysis
3.7.1. Average Shortest Path (L)
3.7.2. Betweenness Centrality (BC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Betweenness centrality |
| CA | Carbonic anhydrase |
| CAH | Carbonic anhydrase |
| CAM | Calmodulin |
| CARP | Cabonic anhydrase-related protein |
| DCC | Dynamic cross correlation |
| DRN | Dynamic residue network |
| MD | Molecular dynamics |
| L | Average shortest path |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| Rg | Radius of gyration |
| SNV | Single nucleotide variation |
| WT | Wild type |
References
- Consortium, U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Picaud, S.S.; Muniz, J.R.; Kramm, A.; Pilka, E.S.; Kochan, G.; Oppermann, U.; Yue, W.W. Crystal structure of human carbonic anhydrase-related protein VIII reveals the basis for catalytic silencing. Proteins Struct. Funct. Bioinform. 2009, 76, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, E.; Wagner, W.; Kapfhammer, J.P. Carbonic anhydrase 8 expression in purkinje cells is controlled by PKCγ activity and regulates purkinje cell dendritic growth. Mol. Neurobiol. 2016, 53, 5149–5160. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.S.; Wang, T.K.; Liu, Y.W.; Li, Y.T.; Chi, T.H.; Chou, C.W.; Hsieh, M. Roles of carbonic anhydrase 8 in neuronal cells and zebrafish. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Nishimori, I.; Takeuchi, T.; Fujikawa-Adachi, K.; Ohtsuki, Y.; Onishi, S. Developmental expression of carbonic anhydrase-related proteins VIII, X, and XI in the human brain. Neuroscience 2002, 112, 93–99. [Google Scholar] [CrossRef]
- Hirota, J.; Hideaki, A.; Hamada, K.; Mikoshiba, K. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem. J. 2003, 372, 435–441. [Google Scholar] [CrossRef]
- Aspatwar, A.; Tolvanen, M.E.; Ortutay, C.; Parkkila, S. Carbonic anhydrase related proteins: Molecular biology and evolution. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Springer: Berlin, Germany, 2014; pp. 135–156. [Google Scholar]
- Aspatwar, A.; Tolvanen, M.E.; Jokitalo, E.; Parikka, M.; Ortutay, C.; Harjula, S.K.E.; Rämet, M.; Vihinen, M.; Parkkila, S. Abnormal cerebellar development and ataxia in CARP VIII morphant zebrafish. Hum. Mol. Genet. 2013, 22, 417–432. [Google Scholar] [CrossRef]
- Jiao, Y.; Yan, J.; Zhao, Y.; Donahue, L.R.; Beamer, W.G.; Li, X.; Roe, B.A.; LeDoux, M.S.; Gu, W. Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics 2005, 171, 1239–1246. [Google Scholar] [CrossRef]
- Zhuang, G.Z.; Keeler, B.; Grant, J.; Bianchi, L.; Fu, E.S.; Zhang, Y.P.; Erasso, D.M.; Cui, J.G.; Wiltshire, T.; Li, Q.; et al. Carbonic anhydrase-8 regulates inflammatory pain by inhibiting the ITPR1-cytosolic free calcium pathway. PLoS ONE 2015, 10, e0118273. [Google Scholar] [CrossRef]
- Levitt, R.C.; Zhuang, G.Z.; Keeler, B.; Grant, J.; Bianchi, L.; Fu, E.S.; Zhang, Y.P.; Erasso, D.M.; Cui, J.G.; Wiltshire, T.; et al. Carbonic anhydrase-8 gene therapy inhibits the ITPR1-cytosolic free calcium pathway producing analgesia and anti-hyperalgesia. Mol. Pain 2014, 10, O7. [Google Scholar] [CrossRef][Green Version]
- Hur, E.M.; Park, Y.S.; Huh, Y.H.; Yoo, S.H.; Woo, K.C.; Choi, B.H.; Kim, K.T. Junctional membrane inositol 1,4,5-trisphosphate receptor complex coordinates sensitization of the silent EGF-induced Ca2+ signaling. J. Cell Biol. 2005, 169, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, E.; Kapfhammer, J.P. Calcium signaling, PKC gamma, IP3R1 and CAR8 link spinocerebellar ataxias and Purkinje cell dendritic development. Curr. Neuropharmacol. 2018, 16, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lamont, M.G.; Weber, J.T. The role of calcium in synaptic plasticity and motor learning in the cerebellar cortex. Neurosci. Biobehav. Rev. 2012, 36, 1153–1162. [Google Scholar] [CrossRef]
- Gruol, D.; Manto, M.; Haines, D. Ca 2+ Signaling in Cerebellar Purkinje Neurons. Cerebellum 2012, 11, 605–608. [Google Scholar] [CrossRef]
- Türkmen, S.; Guo, G.; Garshasbi, M.; Hoffmann, K.; Alshalah, A.J.; Mischung, C.; Kuss, A.; Humphrey, N.; Mundlos, S.; Robinson, P.N. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet. 2009, 5, e1000487. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef]
- Yamada, M.; Miyawaki, A.; Saito, K.; Nakajima, T.; Yamamoto-Hino, M.; Ryo, Y.; Furuichi, T.; Mikoshiba, K. The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. Biochem. J. 1995, 308, 83–88. [Google Scholar] [CrossRef]
- Kasri, N.N.; Török, K.; Galione, A.; Garnham, C.; Callewaert, G.; Missiaen, L.; Parys, J.B.; De Smedt, H. Endogenously bound calmodulin is essential for the function of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 2006, 281, 8332–8338. [Google Scholar] [CrossRef]
- Bosanac, I.; Alattia, J.R.; Mal, T.K.; Chan, J.; Talarico, S.; Tong, F.K.; Tong, K.I.; Yoshikawa, F.; Furuichi, T.; Iwai, M.; et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 2002, 420, 696. [Google Scholar] [CrossRef]
- Bosanac, I.; Yamazaki, H.; Matsu-ura, T.; Michikawa, T.; Mikoshiba, K.; Ikura, M. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol. Cell 2005, 17, 193–203. [Google Scholar] [CrossRef]
- Sienaert, I.; Kasri, N.N.; Vanlingen, S.; Parys, J.B.; Callewaert, G.; Missiaen, L.; De Smedt, H. Localization and function of a calmodulin-apocalmodulin-binding domain in the N-terminal part of the type 1 inositol 1,4,5-trisphosphate receptor. Biochem. J. 2002, 365, 269. [Google Scholar] [CrossRef]
- Hsiao, C.T.; Liu, Y.T.; Liao, Y.C.; Hsu, T.Y.; Lee, Y.C.; Soong, B.W. Mutational analysis of ITPR1 in a Taiwanese cohort with cerebellar ataxias. PLoS ONE 2017, 12, e0187503. [Google Scholar] [CrossRef] [PubMed]
- Parolin Schnekenberg, R.; Perkins, E.M.; Miller, J.W.; Davies, W.I.; D’Adamo, M.C.; Pessia, M.; Fawcett, K.A.; Sims, D.; Gillard, E.; Hudspith, K.; et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 2015, 138, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, T.; Barth, P.; Reneman, L.; Appelhof, B.; Baas, F.; Poll-The, B.T. A de novo missense mutation in the inositol 1,4,5-triphosphate receptor type 1 gene causing severe pontine and cerebellar hypoplasia: Expanding the phenotype of ITPR1-related spinocerebellar ataxia’s. Am. J. Med Genet. Part A 2017, 173, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tang, T.S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J. Neurosci. 2009, 29, 9148–9162. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- OMIM. Online Mendelian Inheritance in Man. 2018. Available online: https://www.omim.org/about (accessed on 15 May 2019).
- Hirasawa, M.; Xu, X.; Trask, R.B.; Maddatu, T.P.; Johnson, B.A.; Naggert, J.K.; Nishina, P.M.; Ikeda, A. Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol. Cell. Neurosci. 2007, 35, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kou, I.; Sato, H.; Emi, M.; Ito, H.; Hosoi, T.; Ikegawa, S. Nucleotide variations in genes encoding carbonic anhydrase 8 and 10 associated with femoral bone mineral density in Japanese female with osteoporosis. J. Bone Miner. Metab. 2009, 27, 213–216. [Google Scholar] [CrossRef]
- Kaya, N.; Aldhalaan, H.; Al-Younes, B.; Colak, D.; Shuaib, T.; Al-Mohaileb, F.; Al-Sugair, A.; Nester, M.; Al-Yamani, S.; Al-Bakheet, A.; et al. Phenotypical spectrum of cerebellar ataxia associated with a novel mutation in the CA8 gene, encoding carbonic anhydrase (CA) VIII. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2011, 156, 826–834. [Google Scholar] [CrossRef]
- Aspatwar, A.; EE Tolvanen, M.; Ortutay, C.; Parkkila, S. Carbonic anhydrase related protein VIII and its role in neurodegeneration and cancer. Curr. Pharm. Des. 2010, 16, 3264–3276. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2017, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.K.; Tastan Bishop, Ö. HUMA: A platform for the analysis of genetic variation in humans. Hum. Mutat. 2018, 39, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins: Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Türkmen, S.; Demirhan, O.; Hoffmann, K.; Diers, A.; Zimmer, C.; Sperling, K.; Mundlos, S. Cerebellar hypoplasia and quadrupedal locomotion in humans as a recessive trait mapping to chromosome 17p. J. Med Genet. 2006, 43, 461–464. [Google Scholar] [CrossRef][Green Version]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- De Vries, S.J.; Bonvin, A.M. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Budagyan, L.; Raush, E.; Totrov, M.; Abagyan, R. PIER: Protein interface recognition for structural proteomics. Proteins Struct. Funct. Bioinform. 2007, 67, 400–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, H.X. Prediction of interface residues in protein–protein complexes by a consensus neural network method: Test against NMR data. Proteins Struct. Funct. Bioinform. 2005, 61, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Neuvirth, H.; Raz, R.; Schreiber, G. ProMate: A structure based prediction program to identify the location of protein–protein binding sites. J. Mol. Biol. 2004, 338, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein–protein interactions. Proteins Struct. Funct. Bioinform. 2007, 66, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhang, C.; Liu, S.; Zhou, Y. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006, 34, 3698–3707. [Google Scholar] [CrossRef] [PubMed]
- Sanyanga, T.A.; Nizami, B.; Tastan Bishop, Ö. Mechanism of Action of Non-Synonymous Single Nucleotide Variations Associated with α-Carbonic Anhydrase II Deficiency. Molecules 2019, 24, 3987. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Supuran, C.T. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin. Drug Discov. 2020, 1–16. [Google Scholar] [CrossRef]
- Sjöblom, B.; Elleby, B.; Wallgren, K.; Jonsson, B.H.; Lindskog, S. Two point mutations convert a catalytically inactive carbonic anhydrase-related protein (CARP) to an active enzyme. FEBS Lett. 1996, 398, 322–325. [Google Scholar] [CrossRef]
- Brown, D.K.; Penkler, D.L.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017, 33, 2768–2771. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Schafmeister, C.; Ross, W.; Romanovski, V. LEaP; University of California: San Francisco, CA, USA, 1995. [Google Scholar]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2018-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNV | ||||||
|---|---|---|---|---|---|---|
| S100A | S100L | S100P | E109D | G162R | R237Q | |
| rs ID | rs267606695 | rs149391728 | rs267606695 | rs7464181 | rs1421833180 | rs387906598 |
| MAF | 0.01 | <0.01 | <0.01 | 0.50 | <0.01 | <0.01 |
| I-Mutant (ΔΔG) | −0.66 | −0.31 | −0.27 | −0.16 | −0.84 | −0.58 |
| MUPro (ΔΔG) | −1.23 | −0.17 | −1.39 | −0.41 | −0.54 | −1.05 |
| Stability | Decrease | Decrease | Decrease | Decrease | Decrease | Decrease |
| Clinical significance | Pathogenic | Benign | Pathogenic | Benign | Pathogenic | Pathogenic |
| z-DOPE score | −1.374 | −1.381 | −1.41 | −1.436 | −1.357 | −1.393 |
| Protein | Metric Mean | % Difference (WT – Variant) |
|---|---|---|
| RMSD | ||
| WT | 2.507 | 0.00 |
| S100A | 2.465 | 1.68 |
| S100L | 2.002 | 20.14 |
| S100P | 2.251 | 10.21 |
| E109D | 2.222 | 11.37 |
| G162R | 2.862 | −14.16 |
| R237Q | 2.248 | 10.33 |
| Rg | ||
| WT | 18.120 | 0.00 |
| S100A | 17.961 | 0.88 |
| S100L | 17.926 | 1.07 |
| S100P | 18.024 | 0.53 |
| E109D | 17.865 | 1.41 |
| G162R | 18.113 | 0.04 |
| R237Q | 17.944 | 0.97 |
| Variant Protein | Dynamic Residue Network | |
|---|---|---|
| Residue Accessibility Increase (Positive L) | Residue Communication Reduction (Positive BC) | |
| S100A | Glu23 Glu24 Glu25 Gly26 Val27 Glu28 Trp29 Val35 Asp43 | Tyr31Glu32Trp37Val40Phe41 Thr88 Val91 Leu93 Lys96 Gly271 |
| S100L | Glu23 Glu25 Gly26 Val27 Glu28 Trp29 Val35 Asp43 | Gly30 Glu33Trp37Val40Phe41 Asp85 His87 Thr88 Lys96 Arg251 |
| S100P | Glu23 Glu24 Glu25 Gly26 Glu28 Trp29 Gly30 Val35 Thr255 | Trp29Gly30 Glu33Trp37Val40Phe41 Lys96 Gly126 Ile224 Pro225 |
| E109D | Glu23 Glu24 Glu25 Gly26 Val27 Glu28 Trp29 Asp43 Leu70 | Tyr31Trp37 Ala44 Lys96 Pro103 Val115 Ile224 Asp272 |
| G162R | Glu23 Glu24 Glu25 Gly26 Val27 Glu28 Val35 Glu36 Leu39 | Gly30Tyr31Glu32Trp37Val40Phe41 Gly86 Val91 Leu93 Val157 Ile224 Trp233 Gly271 |
| R237Q | Glu23 Glu24 Glu25 Gly26 Val27 Trp29 Gly178 | Tyr31Glu32Trp37Phe41Asp85 Thr88 Val91 Lys96 Glu114 Val115 Arg116 Ile224 Leu240 Gly271 |
| Residue Accessibility Decrease (NegativeL) | Residue Communication Increase (NegativeBC) | |
| S100A | Glu32 Lys94 Lys96 | Glu23 Trp29 His87 Ile89 Phe117 Leu253 Arg275 |
| S100L | Glu32 Glu33 Gly34Trp37 Val40 Phe41 Lys96 Val263 Glu264 Gly265 Cys266 Asp267 | Trp29 Glu128 Arg254 Gly268 Leu270 Asn273 Phe274 Arg275 |
| S100P | Trp37Val40Phe41 Lys96 Pro225 Pro226 | Glu28 Ser127 Ile143 Thr255 Leu270 Phe274 Arg275 Pro276 |
| E109D | Tyr31Glu32 Glu33 Lys94 Lys96 | Glu25 Trp29 Gly30 Asp43 Leu70 His87 Ile143 Gly268 |
| G162R | Val157 Ala260 Leu262 Val263 Leu280 | Val35 Glu36 His87 Arg162 Leu168 Ile234 Arg275 Pro276 |
| R237Q | Lys94 Lys96 | Trp29Phe117 Met138 Leu142 His176 Gly178 Leu206 Leu270 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanyanga, T.A.; Tastan Bishop, Ö. Structural Characterization of Carbonic Anhydrase VIII and Effects of Missense Single Nucleotide Variations to Protein Structure and Function. Int. J. Mol. Sci. 2020, 21, 2764. https://doi.org/10.3390/ijms21082764
Sanyanga TA, Tastan Bishop Ö. Structural Characterization of Carbonic Anhydrase VIII and Effects of Missense Single Nucleotide Variations to Protein Structure and Function. International Journal of Molecular Sciences. 2020; 21(8):2764. https://doi.org/10.3390/ijms21082764
Chicago/Turabian StyleSanyanga, Taremekedzwa Allan, and Özlem Tastan Bishop. 2020. "Structural Characterization of Carbonic Anhydrase VIII and Effects of Missense Single Nucleotide Variations to Protein Structure and Function" International Journal of Molecular Sciences 21, no. 8: 2764. https://doi.org/10.3390/ijms21082764
APA StyleSanyanga, T. A., & Tastan Bishop, Ö. (2020). Structural Characterization of Carbonic Anhydrase VIII and Effects of Missense Single Nucleotide Variations to Protein Structure and Function. International Journal of Molecular Sciences, 21(8), 2764. https://doi.org/10.3390/ijms21082764