In Vivo Targets of Pasteurella Multocida Toxin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
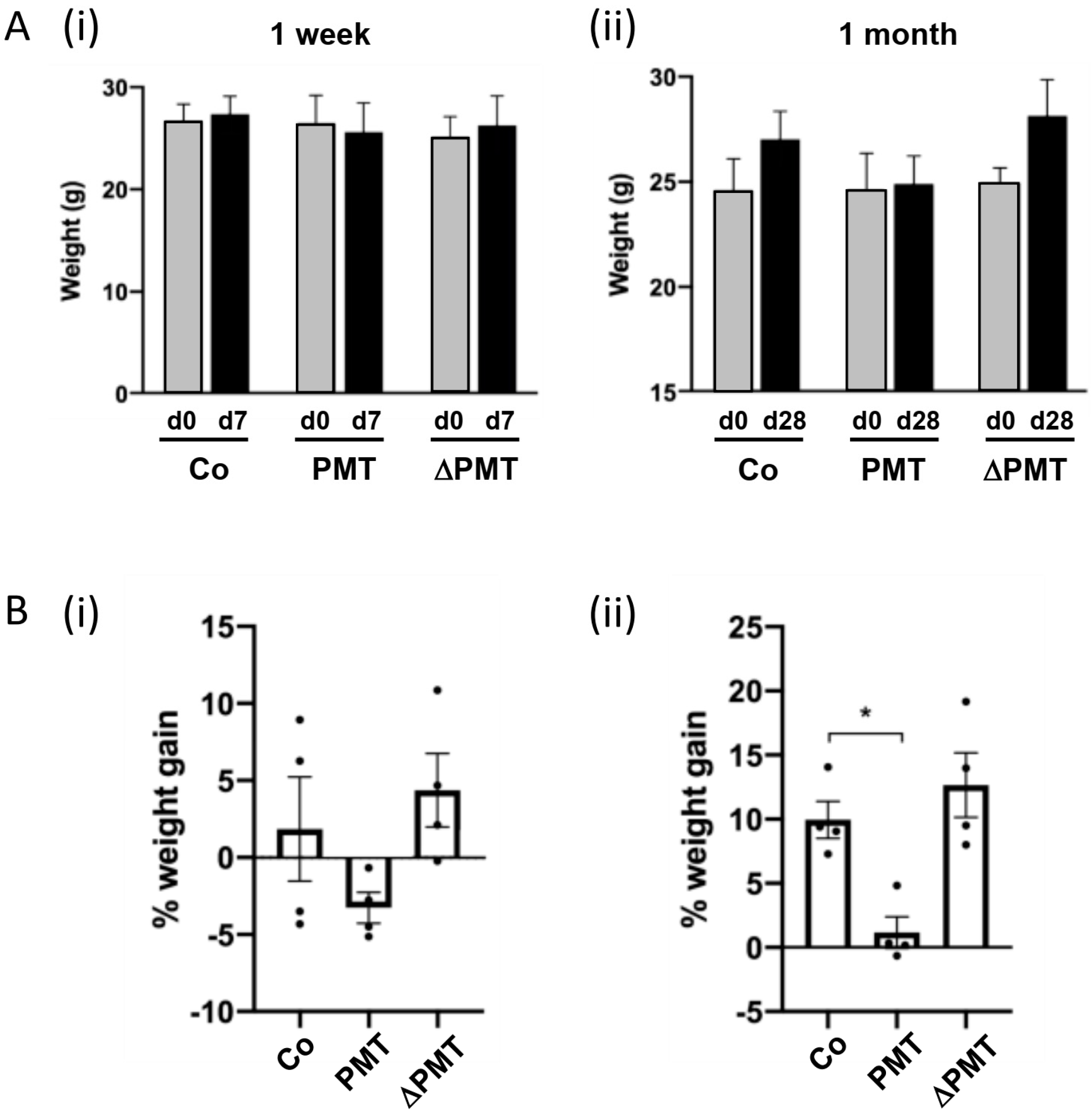
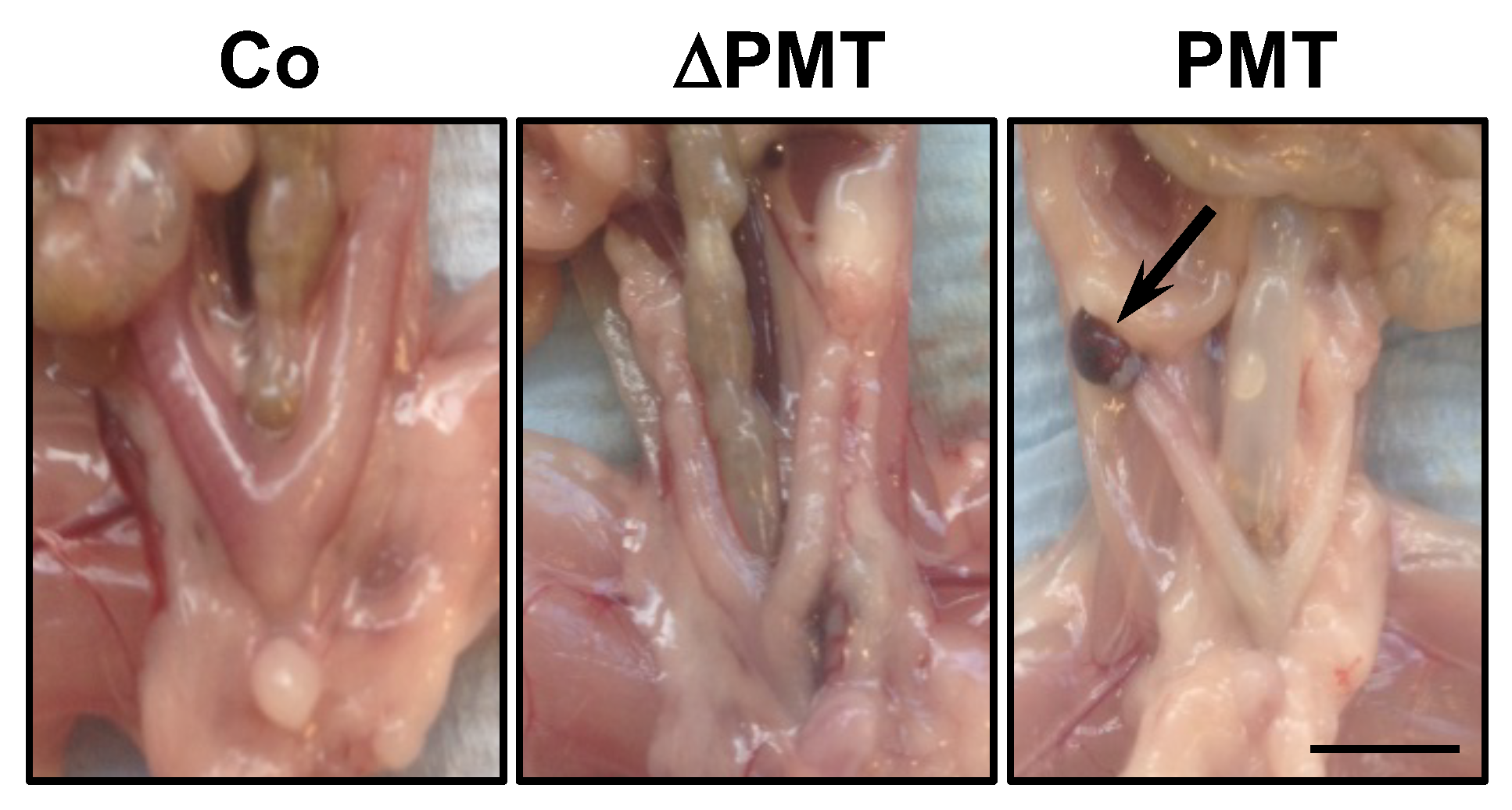
2.1. Effects of PMT Treatment In Vivo
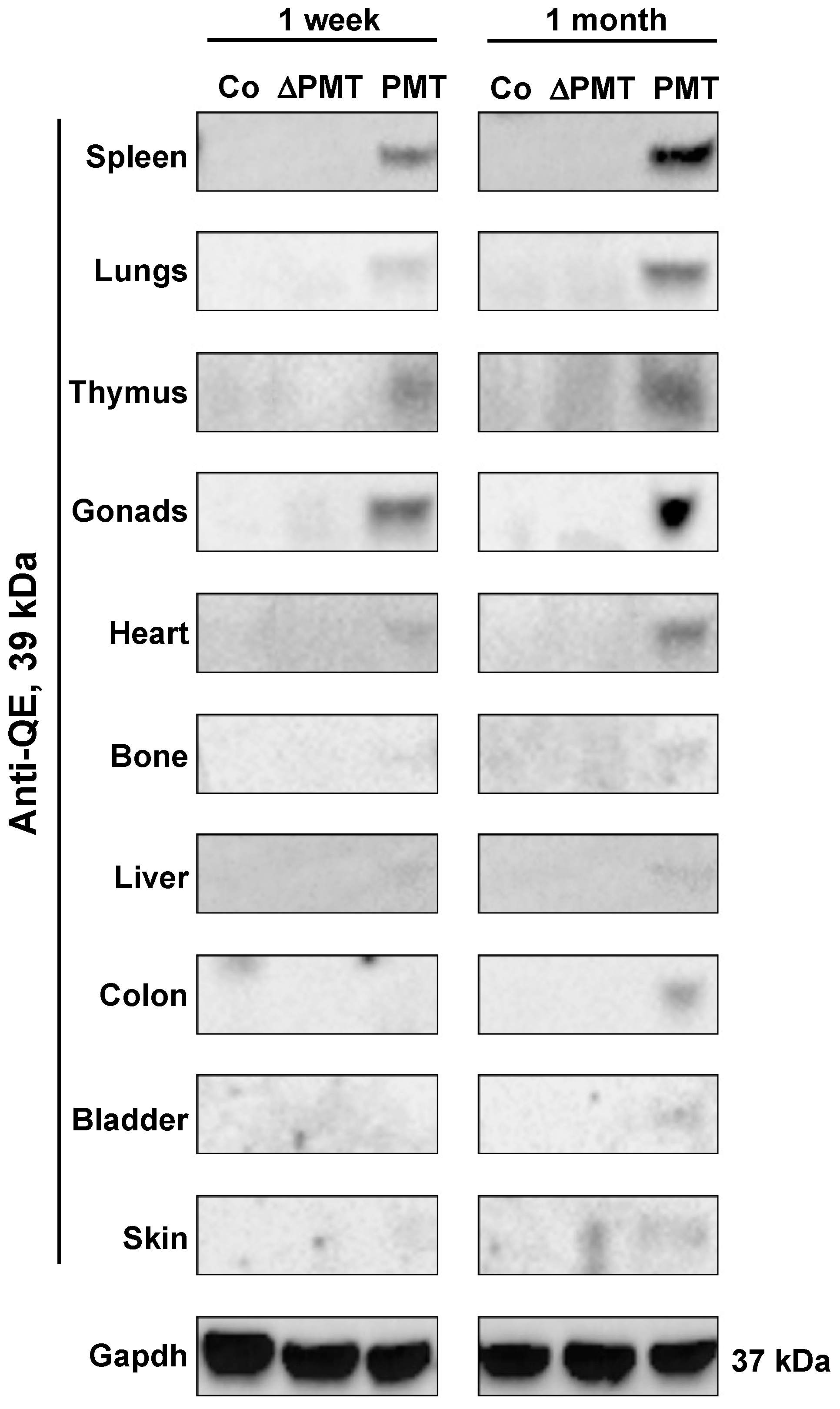
2.2. PMT Modifies G-Proteins In Vivo
2.3. PMT Treatment Induces Cell Proliferation Markers In Vivo
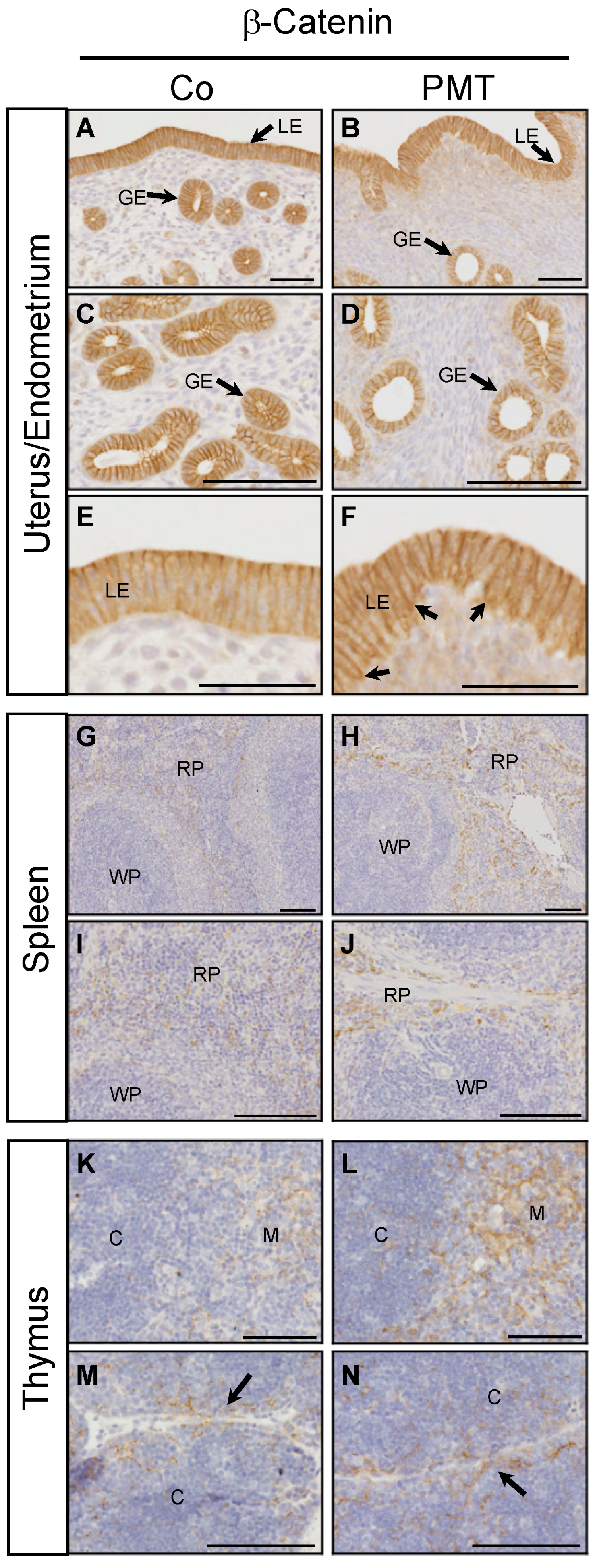
2.4. PMT Treatment Stimulates Active β-Catenin In Vivo
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Histology
4.3. Tissue Homogenisation
4.4. Immunohistochemistry
4.5. Western Blot Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harper, M.; Boyce, J.D.; Adler, B. Pasteurella multocida pathogenesis: 125 years after Pasteur. FEMS Microbiol. Lett. 2006, 265, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.R.; Buker, E.O.; Bell, J.F.; Jellison, W.L. Pasteurella multocida in animals’ mouths. Rocky Mt. Med. J. 1968, 65, 45–46. [Google Scholar] [PubMed]
- Donnio, P.Y.; Avril, J.L.; Andre, P.M.; Vaucel, J. Dermonecrotic toxin production by strains of Pasteurella multocida isolated from man. J. Med. Microbiol. 1991, 34, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Dincman, T.; Clyburn, B.E.; Steed, L.L.; Rockey, D.C. Clinical Features and Outcomes of Pasteurella multocida Infection. Medicine (Baltimore) 2015, 94, e1285. [Google Scholar] [CrossRef] [PubMed]
- Hendrie, J. Pasteurella multocida bacteremia in humans: A clinical report. Can. Fam. Physician 1974, 20, 79–81. [Google Scholar]
- Holst, E.; Rollof, J.; Larsson, L.; Nielsen, J.P. Characterization and distribution of Pasteurella species recovered from infected humans. J. Clin. Microbiol. 1992, 30, 2984–2987. [Google Scholar] [CrossRef]
- Körmöndi, S.; Terhes, G.; Pál, Z.; Varga, E.; Harmati, M.; Buzás, K.; Urbán, E. Human Pasteurellosis Health Risk for Elderly Persons Living with Companion Animals. Emerg. Infect. Dis. 2019, 25, 229–235. [Google Scholar] [CrossRef]
- Escande, F.; Lion, C. Epidemiology of human infections by Pasteurella and related groups in France. Zentralbl. Bakteriol. 1993, 279, 131–139. [Google Scholar] [CrossRef]
- Heddleston, K.L.; Gallagher, J.E.; Rebers, P.A. Fowl cholera: Gel diffusion precipitin test for serotyping Pasteurella multocida from avian species. Avian. Dis. 1972, 16, 925–936. [Google Scholar] [CrossRef]
- Rutter, J.M. Virulence of Pasteurella multocida in atrophic rhinitis of gnotobiotic pigs infected with Bordetella bronchiseptica. Res. Vet. Sci. 1983, 34, 287–295. [Google Scholar] [CrossRef]
- Lax, A.J.; Chanter, N. Cloning of the toxin gene from Pasteurella multocida and its role in atrophic rhinitis. J. Gen. Microbiol. 1990, 136, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Harmey, D.; Stenbeck, G.; Nobes, C.D.; Lax, A.J.; Grigoriadis, A.E. Regulation of osteoblast differentiation by Pasteurella multocida toxin (PMT): A role for Rho GTPase in bone formation. J. Bone Miner Res. 2004, 19, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Siegert, P.; Schmidt, G.; Papatheodorou, P.; Wieland, T.; Aktories, K.; Orth, J.H. Pasteurella multocida toxin prevents osteoblast differentiation by transactivation of the MAP-kinase cascade via the Gα(q/11)--p63RhoGEF--RhoA axis. PLoS Pathog. 2013, 9, e1003385. [Google Scholar] [CrossRef] [PubMed]
- Strack, J.; Heni, H.; Gilsbach, R.; Hein, L.; Aktories, K.; Orth, J.H. Noncanonical G-protein-dependent modulation of osteoclast differentiation and bone resorption mediated by Pasteurella multocida toxin. mBio 2014, 5, e02190. [Google Scholar] [CrossRef]
- Orth, J.H.; Fester, I.; Preuss, I.; Agnoletto, L.; Wilson, B.A.; Aktories, K. Activation of Galpha (i) and subsequent uncoupling of receptor-Galpha(i) signaling by Pasteurella multocida toxin. J. Biol. Chem. 2008, 283, 23288–23294. [Google Scholar] [CrossRef]
- Preuss, I.; Kurig, B.; Nürnberg, B.; Orth, J.H.; Aktories, K. Pasteurella multocida toxin activates Gbetagamma dimers of heterotrimeric G proteins. Cell Signal 2009, 21, 551–558. [Google Scholar] [CrossRef]
- Preuss, I.; Hildebrand, D.; Orth, J.H.; Aktories, K.; Kubatzky, K.F. Pasteurella multocida toxin is a potent activator of anti-apoptotic signalling pathways. Cell Microbiol. 2010, 12, 1174–1185. [Google Scholar] [CrossRef]
- Rozengurt, E.; Higgins, T.; Chanter, N.; Lax, A.J.; Staddon, J.M. Pasteurella multocida toxin: Potent mitogen for cultured fibroblasts. Proc. Natl. Acad. Sci. USA 1990, 87, 123–127. [Google Scholar] [CrossRef]
- Wilson, B.A.; Zhu, X.; Ho, M.; Lu, L. Pasteurella multocida toxin activates the inositol triphosphate signaling pathway in Xenopus oocytes via G(q)alpha-coupled phospholipase C-beta1. J. Biol. Chem. 1997, 272, 1268–1275. [Google Scholar] [CrossRef]
- Orth, J.H.; Lang, S.; Taniguchi, M.; Aktories, K. Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of G{alpha} proteins, G{alpha}q and G{alpha}12/13. J. Biol. Chem. 2005, 280, 36701–36707. [Google Scholar] [CrossRef]
- Orth, J.H.; Fester, I.; Siegert, P.; Weise, M.; Lanner, U.; Kamitani, S.; Tachibana, T.; Wilson, B.A.; Schlosser, A.; Horiguchi, Y.; et al. Substrate specificity of Pasteurella multocida toxin for α subunits of heterotrimeric G proteins. FASEB J. 2013, 27, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Zywietz, A.; Gohla, A.; Schmelz, M.; Schultz, G.; Offermanns, S. Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. involvement of Gq but not G11. J. Biol. Chem. 2001, 276, 3840–3845. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.H.; Lang, S.; Preuss, I.; Milligan, G.; Aktories, K. Action of Pasteurella multocida toxin on Galpha(q) is persistent and independent of interaction with G-protein-coupled receptors. Cell Signal 2007, 19, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Babb, R.C.; Homer, K.A.; Robbins, J.; Lax, A.J. Modification of heterotrimeric G-proteins in Swiss 3T3 cells stimulated with Pasteurella multocida toxin. PLoS ONE 2012, 7, e47188. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.R.; Pullinger, G.D.; Lax, A.J. Pasteurella multocida toxin facilitates inositol phosphate formation by bombesin through tyrosine phosphorylation of G alpha q. J. Biol. Chem. 2003, 278, 32719–32725. [Google Scholar] [CrossRef]
- Kamitani, S.; Ao, S.; Toshima, H.; Tachibana, T.; Hashimoto, M.; Kitadokoro, K.; Fukui-Miyazaki, A.; Abe, H.; Horiguchi, Y. Enzymatic actions of Pasteurella multocida toxin detected by monoclonal antibodies recognizing the deamidated α subunit of the heterotrimeric GTPase Gq. FEBS J. 2011, 278, 2702–2712. [Google Scholar] [CrossRef]
- Banu, A.; Liu, K.J.; Lax, A.J.; Grigoriadis, A.E. G-alpha subunit abundance and activity differentially regulate β-catenin signalling. Mol. Cell Biol. 2019, 39, e00422-18. [Google Scholar]
- Mullan, P.B.; Lax, A.J. Pasteurella multocida toxin is a mitogen for bone cells in primary culture. Infect. Immun. 1996, 64, 959–965. [Google Scholar] [CrossRef]
- Staddon, J.M.; Chanter, N.; Lax, A.J.; Higgins, T.E.; Rozengurt, E. Pasteurella multocida toxin, a potent mitogen, stimulates protein kinase C-dependent and -independent protein phosphorylation in Swiss 3T3 cells. J. Biol Chem. 1990, 265, 11841–11848. [Google Scholar]
- Higgins, T.E.; Murphy, A.C.; Staddon, J.M.; Lax, A.J.; Rozengurt, E. Pasteurella multocida toxin is a potent inducer of anchorage-independent cell growth. Proc. Natl. Acad. Sci. USA 1992, 89, 4240–4244. [Google Scholar] [CrossRef]
- Surguy, S.M.; Duricki, D.A.; Reilly, J.M.; Lax, A.J.; Robbins, J. The actions of Pasteurella multocida toxin on neuronal cells. Neuropharmacology 2014, 77, 9–18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Staddon, J.M.; Barker, C.J.; Murphy, A.C.; Chanter, N.; Lax, A.J.; Michell, R.H.; Rozengurt, E. Pasteurella multocida toxin, a potent mitogen, increases inositol 1,4,5-trisphosphate and mobilizes Ca2+ in Swiss 3T3 cells. J. Biol. Chem. 1991, 266, 4840–4847. [Google Scholar] [PubMed]
- Lax, A.J.; Grigoriadis, A.E. Pasteurella multocida toxin: The mitogenic toxin that stimulates signalling cascades to regulate growth and differentiation. Int. J. Med. Microbiol. 2001, 291, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Blocker, D.; Berod, L.; Fluhr, J.W.; Orth, J.; Idzko, M.; Aktories, K.; Norgauer, J. Pasteurella multocida toxin (PMT) activates RhoGTPases, induces actin polymerization and inhibits migration of human dendritic cells, but does not influence macropinocytosis. Int. Immunol. 2006, 18, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Essler, M.; Hermann, K.; Amano, M.; Kaibuchi, K.; Heesemann, J.; Weber, P.C.; Aepfelbacher, M. Pasteurella multocida toxin increases endothelial permeability via Rho kinase and myosin light chain phosphatase. J. Immunol. 1998, 161, 5640–5646. [Google Scholar] [PubMed]
- Lax, A.J. Opinion: Bacterial toxins and cancer--a case to answer? Nat. Rev. Microbiol. 2005, 3, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Lax, A. The Pasteurella multocida toxin: A new paradigm for the link between bacterial infection and cancer. Curr. Top Microbiol. Immunol. 2012, 361, 131–144. [Google Scholar]
- Wilson, B.A.; Ho, M. Pasteurella multocida Toxin Interaction with Host Cells: Entry and Cellular Effects. In Pasteurella multocida: Molecular Biology, Toxins and Infection; Aktories, K., Orth, J.H.C., Adler, B., Eds.; Springer: Berlin, Heidelberg, 2012; pp. 93–111. [Google Scholar]
- Hoskins, I.C.; Thomas, L.H.; Lax, A.J. Nasal infection with Pasteurella multocida causes proliferation of bladder epithelium in gnotobiotic pigs. Vet. Rec. 1997, 140, 22. [Google Scholar] [CrossRef]
- Rutter, J.M.; Mackenzie, A. Pathogenesis of atrophic rhinitis in pigs: A new perspective. Vet. Rec. 1984, 114, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Cheville, N.F.; Rimler, R.B. A protein toxin from Pasteurella multocida type D causes acute and chronic hepatic toxicity in rats. Vet. Pathol. 1989, 26, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Weise, M.; Vettel, C.; Spiger, K.; Gilsbach, R.; Hein, L.; Lorenz, K.; Wieland, T.; Aktories, K.; Orth, J.H. A systemic Pasteurella multocida toxin aggravates cardiac hypertrophy and fibrosis in mice. Cell Microbiol. 2015, 17, 1320–1331. [Google Scholar] [CrossRef]
- Orth, J.H.; Preuss, I.; Fester, I.; Schlosser, A.; Wilson, B.A.; Aktories, K. Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc. Natl. Acad. Sci. USA 2009, 106, 7179–7184. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.N.; Miles, A.J.; Sumner, I.G.; Thomas, L.H.; Lax, A.J. Activity of the mitogenic Pasteurella multocida toxin requires an essential C-terminal residue. Infect. Immun. 1998, 66, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.M.; Taylor, R.J.; Crighton, W.G.; Robertson, I.B.; Benson, J.A. Epidemiological study of Pasteurella multocida and Bordetella bronchiseptica in atrophic rhinitis. Vet. Rec. 1984, 115, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.B.; New, C.W.; Baker, P.K.; Hogg, A.; Underdahl, N.R. Bordetella bronchiseptica and toxigenic type D Pasteurella multocida as agents of severe atrophic rhinitis of swine. Vet. Microbiol. 1987, 13, 179–187. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Barfod, K. The aetiological significance of Bordetella bronchiseptica and Pasteurella multocida in atrophic rhinitis of swine. Nord. Vet. Med. 1981, 33, 513–522. [Google Scholar] [PubMed]
- Ackermann, M.R.; Tappe, J.P., Jr.; Thurston, J.R.; Rimler, R.B.; Shuster, D.E.; Cheville, N.F. Light microscopic and ultrastructural pathology of seminiferous tubules of rats given multiple doses of Pasteurella multocida group D protein toxin. Toxicol. Pathol. 1992, 20, 103–111. [Google Scholar] [CrossRef][Green Version]
- Dominick, M.A.; Rimler, R.B. Turbinate atrophy in gnotobiotic pigs intranasally inoculated with protein toxin isolated from type D Pasteurella multocida. Am. J. Vet. Res. 1986, 47, 1532–1536. [Google Scholar]
- Brothers, M.C.; Ho, M.; Maharjan, R.; Clemons, N.C.; Bannai, Y.; Waites, M.A.; Faulkner, M.J.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Blanke, S.R.; et al. Membrane interaction of Pasteurella multocida toxin involves sphingomyelin. FEBS J. 2011, 278, 4633–4648. [Google Scholar] [CrossRef]
- Chen, B.; Leverette, R.D.; Schwinn, D.A.; Kwatra, M.M. Human G(alpha q): cDNA and tissue distribution. Biochim Biophys Acta 1996, 1281, 125–128. [Google Scholar] [CrossRef][Green Version]
- Jeong, J.W.; Lee, H.S.; Franco, H.L.; Broaddus, R.R.; Taketo, M.M.; Tsai, S.Y.; Lydon, J.P.; DeMayo, F.J. Beta-catenin mediates glandular formation and dysregulation of beta-catenin induces hyperplasia formation in the murine uterus. Oncogene 2009, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Kiewisz, J.; Wasniewski, T.; Kmiec, Z. Participation of WNT and β-Catenin in Physiological and Pathological Endometrial Changes: Association with Angiogenesis. Biomed Res. Int. 2015, 2015, 854056. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Burgy, O.; Königshoff, M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018, 68–69, 67–80. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Cavallo, R.; Dooijes, D.; van Beest, M.; van Es, J.; Loureiro, J.; Ypma, A.; Hursh, D.; Jones, T.; Bejsovec, A.; et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 1997, 88, 789–799. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banu, A.; Lax, A.J.; Grigoriadis, A.E. In Vivo Targets of Pasteurella Multocida Toxin. Int. J. Mol. Sci. 2020, 21, 2739. https://doi.org/10.3390/ijms21082739
Banu A, Lax AJ, Grigoriadis AE. In Vivo Targets of Pasteurella Multocida Toxin. International Journal of Molecular Sciences. 2020; 21(8):2739. https://doi.org/10.3390/ijms21082739
Chicago/Turabian StyleBanu, Arshiya, Alistair J. Lax, and Agamemnon E. Grigoriadis. 2020. "In Vivo Targets of Pasteurella Multocida Toxin" International Journal of Molecular Sciences 21, no. 8: 2739. https://doi.org/10.3390/ijms21082739
APA StyleBanu, A., Lax, A. J., & Grigoriadis, A. E. (2020). In Vivo Targets of Pasteurella Multocida Toxin. International Journal of Molecular Sciences, 21(8), 2739. https://doi.org/10.3390/ijms21082739