Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response—What is First?

 ,
,  ,
,  , , , ,
, , , ,
Abstract
1. Introduction
2. Results
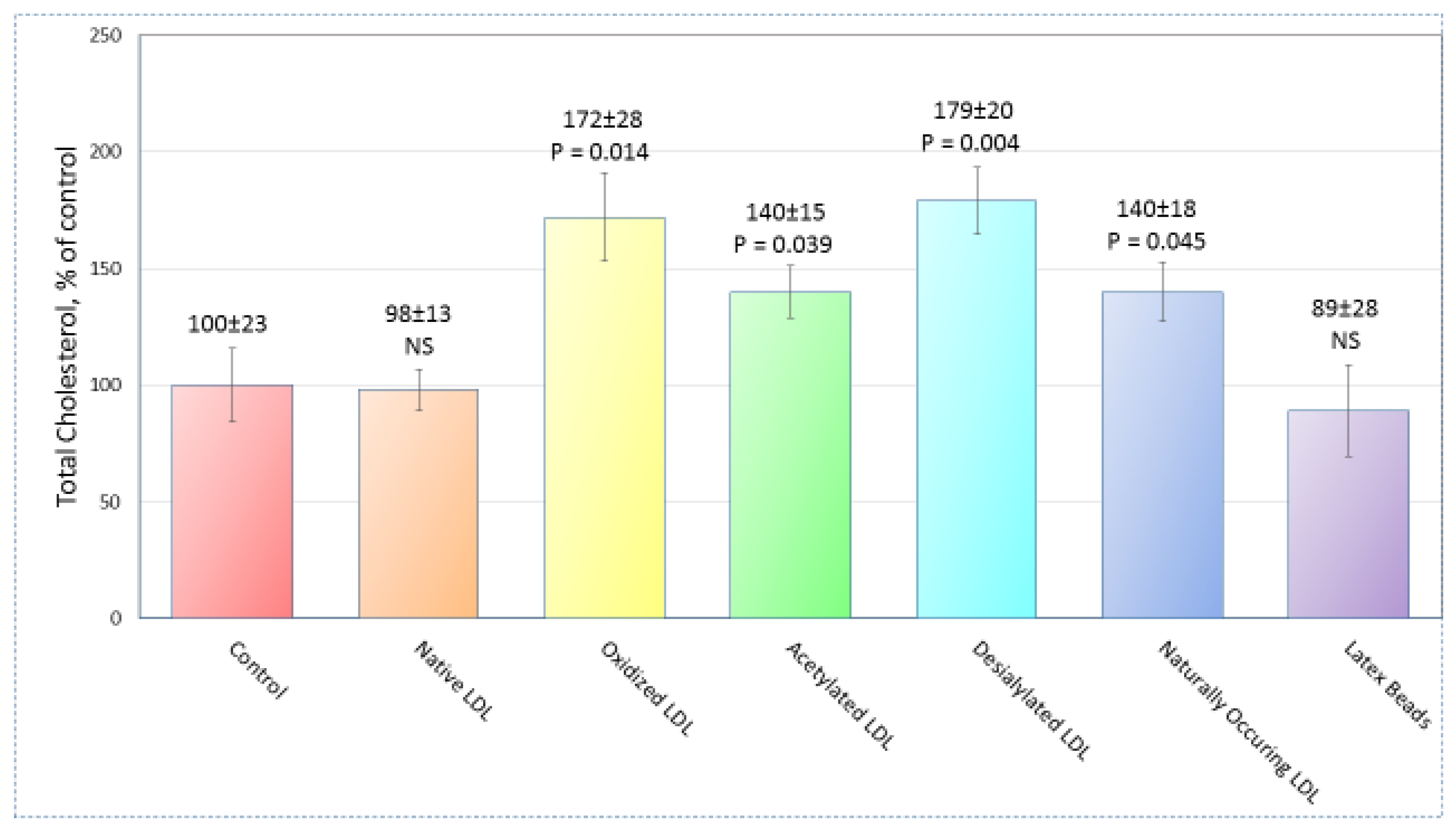
2.1. Sample Preparation
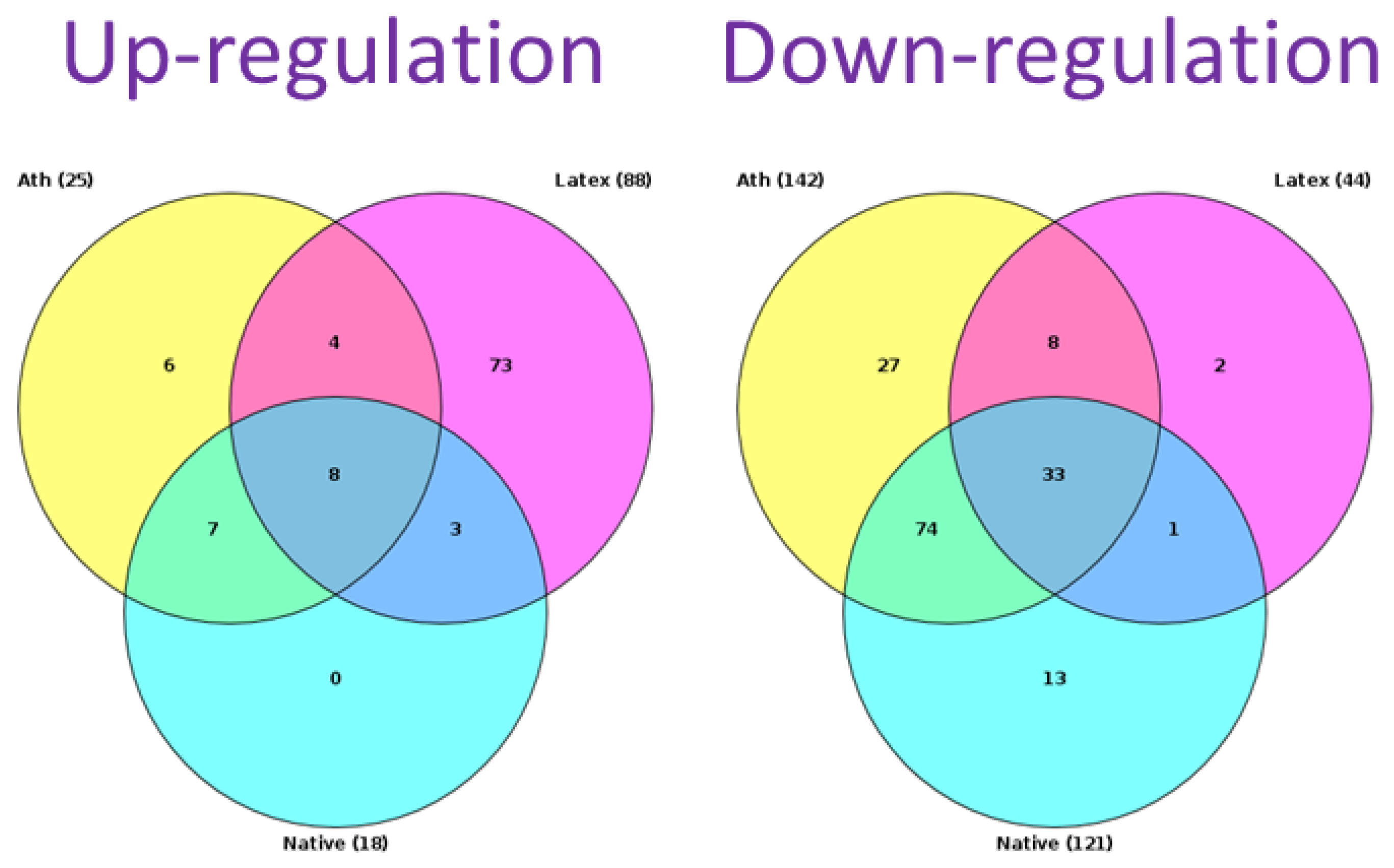
2.2. Search for Signaling Pathways
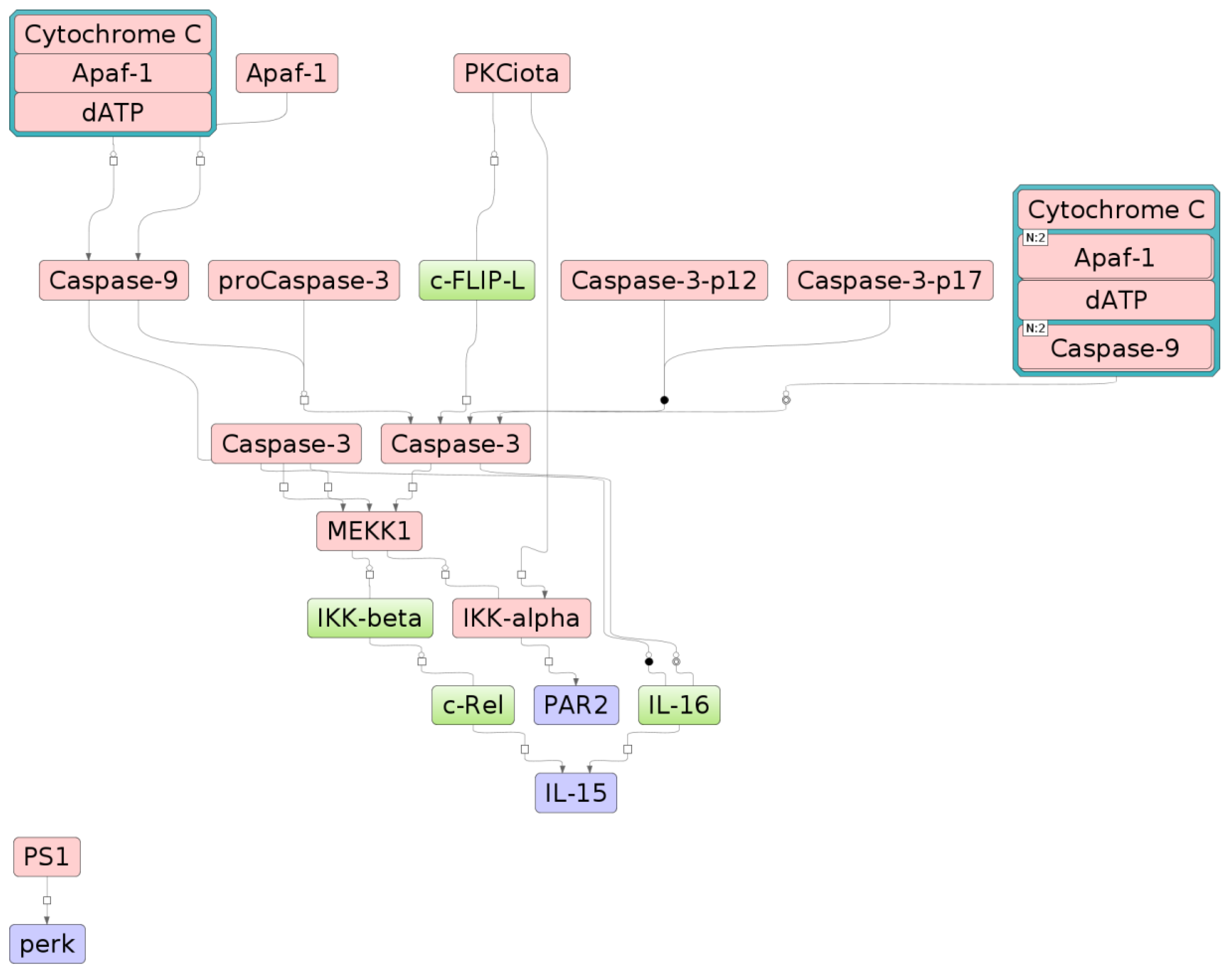
2.3. The Relationship between Key Genes and Signaling Pathways
2.4. Knockdown of Key Genes
3. Discussion
4. Materials and Methods
4.1. Lipoproteins and Latex Beads
4.2. Monocyte-derived Macrophages
4.3. Measurement of Intracellular Cholesterol
4.4. TRANSFAC and TRANSPATH Databases
4.5. Analysis of Enriched Transcription Factor Binding Sites in Promoters of Differentially Expressed Genes
4.6. Identification of Signal Transduction Network
4.7. Gene Expression Knockdown by Small Interference RNA (siRNA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pryma, C.S.; Ortega, C.; Dubland, J.A.; Francis, G.A. Pathways of smooth muscle foam cell formation in atherosclerosis. Curr. Opin. Lipidol. 2019, 30, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N. LDL and foam cell formation as the basis of atherogenesis. Curr. Opin. Lipidol. 2018, 29, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Doodnauth, S.A.; Grinstein, S.; Maxson, M.E. Constitutive and stimulated macropinocytosis in macrophages: Roles in immunity and in the pathogenesis of atherosclerosis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2019, 374, 20180147. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, M.; Hosseini-Fard, R.; Najafi, M. Circulating low density lipoprotein (LDL). Horm. Mol. Biol. Clin. Investig. 2018, 35, 20180024. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, R.; Masana, L. Review of the scientific evolution of gene therapy for the treatment of homozygous familial hypercholesterolaemia: Past, present and future perspectives. J. Med. Genet. 2019, 56, 711–717. [Google Scholar] [CrossRef]
- Jiang, L.; Sun, L.Y.; Dai, Y.F.; Yang, S.W.; Zhang, F.; Wang, L.Y. The distribution and characteristics of LDL receptor mutations in China: A systematic review. Sci. Rep. 2015, 5, 17272. [Google Scholar] [CrossRef]
- Niazi, M.; Galehdar, N.; Jamshidi, M.; Mohammadi, R.; Moayyedkazemi, A. A review of the role of statins in heart failure treatment. Curr. Clin. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Arnold, K.; Arnhold, J.; Zschörnig, O.; Wiegel, D.; Krumbiegel, M. Characterization of chemical modifications of surface properties of low-density lipoproteins. Biomed. Biochim. Acta 1989, 48, 735–742. [Google Scholar]
- Desrumaux, C.; Athias, A.; Masson, D.; Gambert, P.; Lallemant, C.; Lagrost, L. Influence of the electrostatic charge of lipoprotein particles on the activity of the human plasma phospholipid transfer protein. J. Lipid Res. 1998, 39, 131–142. [Google Scholar]
- Fogelman, A.M.; Shechter, I.; Seager, J.; Hokom, M.; Child, J.S.; Edwards, P.A. Malondialdehyde alteration of low-density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc. Natl. Acad. Sci. USA 1980, 77, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Ho, Y.K.; Basu, S.K.; Brown, M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low-density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA 1979, 76, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.E.; Olch, C.L.; Folgelman, A.M. Role of lysines in mediating interaction of modified low-density lipoproteins with the scavenger receptor of human monocyte macrophages. J. Biol. Chem. 1984, 259, 11305–11311. [Google Scholar] [PubMed]
- Steinbrecher, U.P.; Parthasarathy, S.; Leake, D.S.; Witztum, J.L.; Steinberg, D. Modification of low-density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low-density lipoprotein phospholipids. Proc. Natl. Acad. Sci. USA 1984, 81, 3883–3887. [Google Scholar] [CrossRef] [PubMed]
- Vanderyse, L.; Devreese, A.M.; Baert, J.; Vanloo, B.; Lins, L.; Ruysschaert, J.M.; Rosseneu, M. Structural and functional properties of apolipoprotein B in chemically modified low density lipoproteins. Atherosclerosis 1992, 97, 187–199. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A. Modified and dysfunctional lipoproteins in atherosclerosis: Effectors or biomarkers? Curr. Med. Chem. 2019, 26, 1512–1524. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A. Modified lipoproteins as biomarkers of atherosclerosis. Front. Biosci. 2018, 23, 1422–1444. [Google Scholar] [CrossRef]
- McLaren, J.E.; Michael, D.R.; Ashlin, T.G.; Ramji, D.P. Cytokines, macrophage lipid metabolism and foam cells: Implications for cardiovascular disease therapy. Prog. Lipid Res. 2011, 50, 331–347. [Google Scholar] [CrossRef]
- Tertov, V.V.; Orekhov, A.N.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Yaroslavov, A.A.; Smirnov, V.N. Three types of naturally occurring modified lipoproteins induce intracellular lipid accumulation due to lipoprotein aggregation. Circ. Res. 1992, 71, 218–228. [Google Scholar] [CrossRef]
- Kel, A.E.; Stegmaier, P.; Valeev, T.; Koschmann, J.; Poroikov, V.; Kel-Margoulis, O.V.; Wingender, E. Multi-omics “upstream analysis” of regulatory genomic regions helps identifying targets against methotrexate resistance of colon cancer. EuPA Open Proteom. 2016, 13, 1–13. [Google Scholar] [CrossRef]
- Kel, A. Data on master regulators and transcription factor binding sites found by upstream analysis of multi-omics data on methotrexate resistance of colon cancer. Data Brief. 2016, 10, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis 1997, 135, 19–27. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Orekhov, A.N. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem. Biophys. Res. Commun. 1989, 163, 489–494. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified LDL particles activate inflammatory pathways in monocyte-derived macrophages: Transcriptome analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef]
- Kel, A.; Voss, N.; Valeev, T.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E. ExPlain: Finding upstream drug targets in disease gene regulatory networks. SAR QSAR Environ. Res. 2008, 19, 481–494. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef]
- Guthrie, L.N.; Abiraman, K.; Plyler, E.S.; Sprenkle, N.T.; Gibson, S.A.; McFarland, B.C.; Rajbhandari, R.; Rowse, A.L.; Benveniste, E.N.; Meares, G.P. Attenuation of PKR-like ER kinase (PERK) signaling Selectively controls endoplasmic reticulum stress-induced inflammation without compromising immunological responses. J. Biol. Chem. 2016, 291, 15830–15840. [Google Scholar] [CrossRef]
- Chiu, W.K.; Fann, M.; Weng, N.P. Generation and growth of CD28nullCD8+ memory T cells mediated by IL-15 and its induced cytokines. J. Immunol. 2006, 177, 7802–7810. [Google Scholar] [CrossRef]
- Cepero-Donates, Y.; Lacraz, G.; Ghobadi, F.; Rakotoarivelo, V.; Orkhis, S.; Mayhue, M.; Chen, Y.G.; Rola-Pleszczynski, M.; Menendez, A.; Ilangumaran, S.; et al. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine 2016, 82, 102–111. [Google Scholar] [CrossRef]
- Johansson, U.; Lawson, C.; Dabare, M.; Syndercombe-Court, D.; Newland, A.C.; Howells, G.L.; Macey, M.G. Human peripheral blood monocytes express protease receptor-2 and respond to receptor activation by production of IL-6, IL-8, and IL-1β. J. Leukoc. Biol. 2005, 78, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Badeanlou, L.; Furlan-Freguia, C.; Yang, G.; Ruf, W.; Samad, F. Tissue factor-protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation. Nat. Med. 2011, 17, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Tertov, V.V.; Kudryashov, S.A.; Smirnov, V.N. Triggerlike stimulation of cholesterol accumulation and DNA and extracellular matrix synthesis induced by atherogenic serum or low-density lipoprotein in cultured cells. Circ. Res. 1990, 66, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The atherogenic role of circulating modified lipids in atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3651. [Google Scholar] [CrossRef]
- Arnao, V.; Tuttolomondo, A.; Daidone, M.; Pinto, A. Lipoproteins in atherosclerosis process. Curr. Med. Chem. 2019, 26, 1525–1543. [Google Scholar] [CrossRef]
- Ku, G.; Thomas, C.E.; Akeson, A.L.; Jackson, R.L. Induction of interleukin 1 beta expression from human peripheral blood monocyte-derived macrophages by 9-hydroxyoctadecadienoic acid. J. Biol. Chem. 1992, 267, 14183–14188. [Google Scholar]
- Thomas, C.E.; Jackson, R.L.; Ohlweiler, D.F.; Ku, G. Multiple lipid oxidation products in low density lipoproteins induce interleukin-1 beta release from human blood mononuclear cells. J. Lipid Res. 1994, 35, 417–427. [Google Scholar]
- Miller, Y.I.; Viriyakosol, S.; Worrall, D.S.; Boullier, A.; Butler, S.; Witztum, J.L. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1213–1219. [Google Scholar] [CrossRef]
- Wiesner, P.; Choi, S.H.; Almazan, F.; Benner, C.; Huang, W.; Diehl, C.J.; Gonen, A.; Butler, S.; Witztum, J.L.; Glass, C.K.; et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ. Res. 2010, 107, 56–65. [Google Scholar] [CrossRef]
- Gowdar, S.; Syal, S.; Chhabra, L. Probable protective role of diabetes mellitus in takotsubo cardiomyopathy: A review. Vessel Plus. 2017, 1, 129–136. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, X.J.; Cao, L.J.; Liu, X.H.; Liu, Z.H.; Wang, X.Q.; Chen, Q.J.; Lu, L.; Shen, W.F.; Liu, Y. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS ONE 2014, 9, e95935. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Strassheim, D.; Karoor, V.; Stenmark, K.; Verin, A.; Gerasimovskaya, E. A current view of G protein-coupled receptor-mediated signaling in pulmonary hypertension: Finding opportunities for therapeutic intervention. Vessel Plus. 2018, 2, 29. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Andreeva, E.R.; Bobryshev, Y.V. Cellular mechanisms of human atherosclerosis: Role of cell-to-cell communications in subendothelial cell functions. Tissue Cell. 2016, 48, 25–34. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Bobryshev, Y.V.; Korobov, G.A.; Borodachev, E.N.; Postnov, A.Y.; Orekhov, A.N. Quantitative analysis of the expression of caspase 3 and caspase 9 in different types of atherosclerotic lesions in the Hum. aorta. Exp. Mol. Pathol. 2015, 99, 1–6. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.G.; Ivanova, E.A.; Sobenin, I.A. Possible role of mitochondrial DNA mutations in chronification of inflammation: Focus on atherosclerosis. J. Clin. Med. 2020, 9, 978. [Google Scholar] [CrossRef]
- Guo, C.; Ma, R.; Liu, X.; Chen, T.; Li, Y.; Yu, Y.; Duan, J.; Zhou, X.; Li, Y.; Sun, Z. Silica nanoparticles promote oxLDL-induced macrophage lipid accumulation and apoptosis via endoplasmic reticulum stress signaling. Sci. Total Environ. 2018, 631–632, 570–579. [Google Scholar] [CrossRef]
- van Es, T.; van Puijvelde, G.H.; Michon, I.N.; van Wanrooij, E.J.; de Vos, P.; Peterse, N.; van Berkel, T.J.; Kuiper, J. IL-15 aggravates atherosclerotic lesion development in LDL receptor deficient mice. Vaccine 2011, 29, 976–983. [Google Scholar] [CrossRef]
- Lacraz, G.; Rakotoarivelo, V.; Labbé, S.M.; Vernier, M.; Noll, C.; Mayhue, M.; Stankova, J.; Schwertani, A.; Grenier, G.; Carpentier, A.; et al. Deficiency of interleukin-15 confers resistance to obesity by diminishing inflammation and enhancing the thermogenic function of adipose tissues. PLoS ONE 2016, 11, e0162995. [Google Scholar]
- Hara, T.; Phuong, P.T.; Fukuda, D.; Yamaguchi, K.; Murata, C.; Nishimoto, S.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; et al. Protease-activated receptor-2 plays a critical role in vascular inflammation and atherosclerosis in apolipoprotein E-deficient mice. Circulation 2018, 138, 1706–1719. [Google Scholar] [CrossRef] [PubMed]
- Girard, D.; Paquet, M.E.; Paquin, R.; Beaulieu, A.D. Differential effects of interleukin-15 (IL-15) and IL-2 on human neutrophils: Modulation of phagocytosis, cytoskeleton rearrangement, gene expression, and apoptosis by IL-15. Blood 1996, 88, 3176–3184. [Google Scholar] [CrossRef] [PubMed]
- Singha, A.K.; Sarkar, C.; Majumder, D.; Debnath, R.; Saha, M.; Maiti, D. IL-15 and GM-CSF stimulated macrophages enhances phagocytic activity in ENU induced leukemic mice. Immunobiology 2019, 4, 151894. [Google Scholar] [CrossRef]
- Kim, Y.C.; Shin, J.E.; Lee, S.H.; Chung, W.J.; Lee, Y.S.; Choi, B.K.; Choi, Y. Membrane-bound proteinase 3 and PAR2 mediate phagocytosis of non-opsonized bacteria in human neutrophils. Mol. Immunol. 2011, 48, 1966–1974. [Google Scholar] [CrossRef]
- Moraes, T.J.; Martin, R.; Plumb, J.D.; Vachon, E.; Cameron, C.M.; Danesh, A.; Kelvin, D.J.; Ruf, W.; Downey, G.P. Role of PAR2 in murine pulmonary pseudomonal infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, 368–377. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Mikhailenko, I.A. Modification of low-density lipoprotein by desialylation causes lipid accumulation in cultured cells: Discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem. Biophys. Res. Commun. 1989, 162, 206–211. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N. Desialylated low density lipoprotein--naturally occurring modified lipoprotein with atherogenic potency. Atherosclerosis 1991, 86, 153–161. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N.; Jaakkola, O.; Solakivi, T.; Nikkari, T. Characteristics of low-density lipoprotein isolated from circulating immune complexes. Atherosclerosis 1996, 122, 191–199. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Jaakkola, O.; Solakivi, T.; Nikkari, T.; Smirnov, V.N.; Orekhov, A.N. Multiple-modified desialylated low-density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab. Investig. 1992, 67, 665–675. [Google Scholar] [PubMed]
- Tertov, V.V.; Orekhov, A.N.; Sobenin, I.A.; Morrisett, J.D.; Gotto, A.M., Jr.; Guevara, J.G., Jr. Carbohydrate composition of protein and lipid components in sialic acid-rich and -poor low-density lipoproteins from subjects with and without coronary artery disease. J. Lipid Res. 1993, 34, 365–375. [Google Scholar] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Gamble, W.; Vaughan, M.; Kruth, H.S.; Avigan, J. Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J. Lipid Res. 1978, 19, 1068–1070. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, 108–110. [Google Scholar] [CrossRef]
- Krull, M.; Pistor, S.; Voss, N.; Kel, A.; Reuter, I.; Kronenberg, D.; Michael, H.; Schwarzer, K.; Potapov, A.; Choi, C.; et al. TRANSPATH: An information resource for storing and visualizing signaling pathways and their pathological aberrations. Nucleic Acids Res. 2006, 34, 546–551. [Google Scholar] [CrossRef]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef]
- Waleev, T.; Shtokalo, D.; Konovalova, T.; Voss, N.; Cheremushkin, E.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E.; Kel, A. Composite module analyst: Identification of transcription factor binding site combinations using genetic algorithm. Nucleic Acids Res. 2006, 34, 541–545. [Google Scholar] [CrossRef]
- Koschmann, J.; Bhar, A.; Stegmaier, P.; Kel, A.E.; Wingender, E. “Upstream analysis”: An integrated promoter-pathway analysis approach to causal interpretation of microarray data. Microarrays 2015, 4, 270–286. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Regulation | Signaling Pathway |
|---|---|---|
| Naturally Occurring | up | neurotrophic signaling TLR2-mediated signaling TLR9 pathway VEGF-A pathway |
| down | Aurora-B cell cycle regulation Cdc20 deubiquitination Cdc20 ubiquitination cyclinB1 ubiquitination ---> anaphase onset Fzr1 ---> cyclin B1 degradation Metaphase to Anaphase transition securin degradation Usp44 ---> Cdc20 | |
| Desialylation | up | Apo2L pathway Fas pathway insulin ---> ERK neurotrophic signaling TLR9 pathway |
| down | Aurora-B cell cycle regulation Cdc20 deubiquitination Cdc20 ubiquitination cyclinB1 ubiquitination ---> anaphase onset Fzr1 ---> cyclin B1 degradation Metaphase to Anaphase transition securin degradation Usp44 ---> Cdc20 | |
| Acetylation | up | alpha IIb beta3 ---> Rac1 alpha IIb beta3 pathway angiotensin II ---> DAG, CaMKII Aurora-A activation, substrates, and degradation Aurora-A cell cycle regulation B-cell antigen receptor pathway BCR ---> ERK BDNF ---trkB---> MAPK cascade beta-glucan ---> AKT-1 Fas pathway G-alpha-q ---> arachidonic acid, ERK insulin ---> ERK insulin pathway insulin ---Shc---> MAPK cascade KSR scaffold complex neurotensin pathway neurotrophic signaling p38 pathway p53 pathway PDGF A ---> ERK PDGF B ---> ERK PKC ---> ERK1, ERK2 POSH ---> JNK1, JNK2 PRL ---Src, FAK1---> ERK RANKL ---> p38 RANKL pathway Tiam1 ---> p38alpha TLR2 ---Rac1--->AKT TLR2-mediated signaling TLR3 pathway TLR9 pathway tuberin pathway VEGF-A pathway |
| down | Aurora-B cell cycle regulation | |
| Oxidation | up | AR pathway Fas pathway HIF-1alpha pathway IRAK-1 ---MKK3---> TNF p38 pathway p53 pathway PKC ---> ERK1, ERK2 RANKL ---> p38 RANKL pathway TLR2-mediated signaling TLR3 pathway TLR9 pathway |
| down | - |
| Knock-downed Gene | Number of Experiments | Relative Content of Intracellular Cholesterol (SD) | p (t-test) | p (Wilcoxon–Mann–Whitney test) | |||
|---|---|---|---|---|---|---|---|
| vs. ‘Control’ | vs. ‘+ LDL’ | vs ‘Control’ | vs. ‘+ LDL’ | ||||
| EIF2AK3 (-) | Control | 6 | 1.00 ± 0.06 (0.35) | - | - | - | |
| + LDL | 6 | 1.36 ± 0.09 (0.58) | 0.002 | - | 0.001 | - | |
| EIF2AK3 (-) + LDL | 6 | 0.98 ± 0.07 (0.43) | 0.86 NS | 0.002 | 0.53 NS | 0.007 | |
| IL15 | Control | 4 | 1.00 ± 0.10 (0.58) | - | - | - | |
| + LDL | 4 | 1.29 ± 0.07 (0.19) | 0.038 | - | 0.038 | - | |
| IL15 (-) + LDL | 4 | 0.89 ± 0.09 (0.49) | 0.35 NS | 0.031 | 0.35 NS | 0.011 | |
| F2RL1 | Control | 4 | 1.00 ± 0.07 (0.37) | - | - | - | |
| + LDL | 4 | 1.29 ± 0.09 (0.46) | 0.014 | - | 0.016 | - | |
| F2RL1 (-) + LDL | 4 | 1.37 ± 0.13 (0.71) | 0.018 | 0.64 NS | 0.019 | 0.52 NS | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orekhov, A.N.; Sukhorukov, V.N.; Nikiforov, N.G.; Kubekina, M.V.; Sobenin, I.A.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; Kel, A.; et al. Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response—What is First? Int. J. Mol. Sci. 2020, 21, 2716. https://doi.org/10.3390/ijms21082716
Orekhov AN, Sukhorukov VN, Nikiforov NG, Kubekina MV, Sobenin IA, Foxx KK, Pintus S, Stegmaier P, Stelmashenko D, Kel A, et al. Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response—What is First? International Journal of Molecular Sciences. 2020; 21(8):2716. https://doi.org/10.3390/ijms21082716
Chicago/Turabian StyleOrekhov, Alexander N., Vasily N. Sukhorukov, Nikita G. Nikiforov, Marina V. Kubekina, Igor A. Sobenin, Kathy K. Foxx, Sergey Pintus, Philip Stegmaier, Daria Stelmashenko, Alexander Kel, and et al. 2020. "Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response—What is First?" International Journal of Molecular Sciences 21, no. 8: 2716. https://doi.org/10.3390/ijms21082716
APA StyleOrekhov, A. N., Sukhorukov, V. N., Nikiforov, N. G., Kubekina, M. V., Sobenin, I. A., Foxx, K. K., Pintus, S., Stegmaier, P., Stelmashenko, D., Kel, A., Poznyak, A. V., Wu, W.-K., Kasianov, A. S., Makeev, V. Y., Manabe, I., & Oishi, Y. (2020). Signaling Pathways Potentially Responsible for Foam Cell Formation: Cholesterol Accumulation or Inflammatory Response—What is First? International Journal of Molecular Sciences, 21(8), 2716. https://doi.org/10.3390/ijms21082716