Skeletal Dysplasias Caused by Sulfation Defects


Abstract
1. Introduction
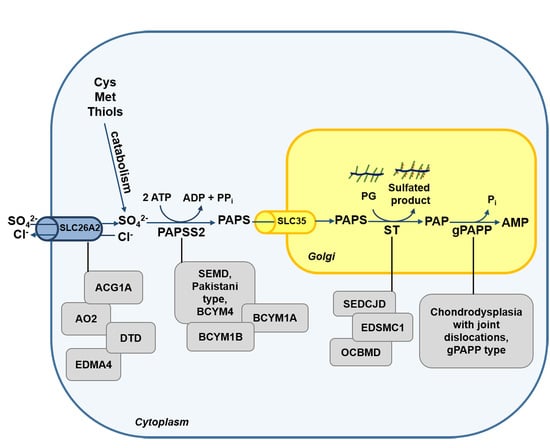
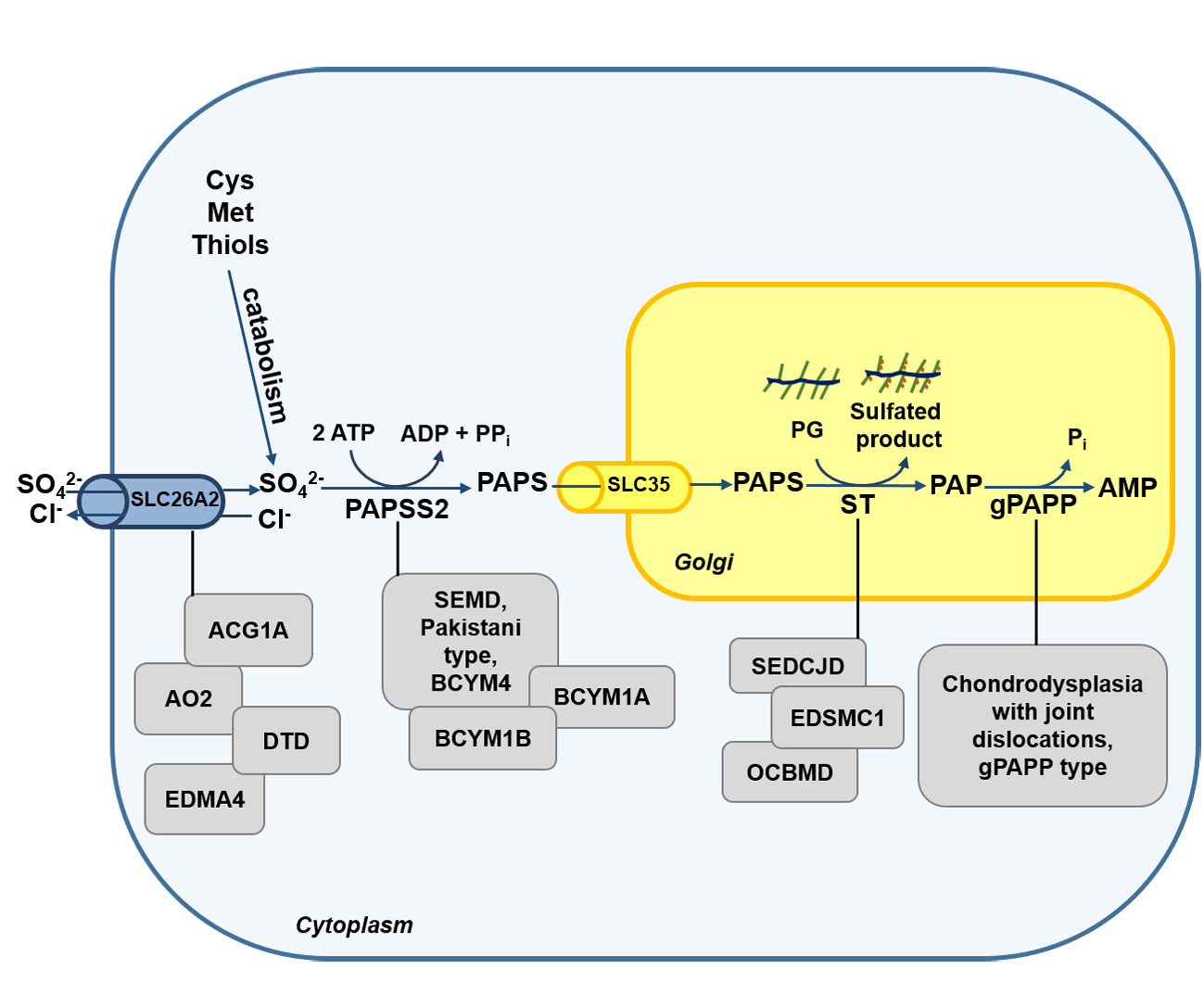
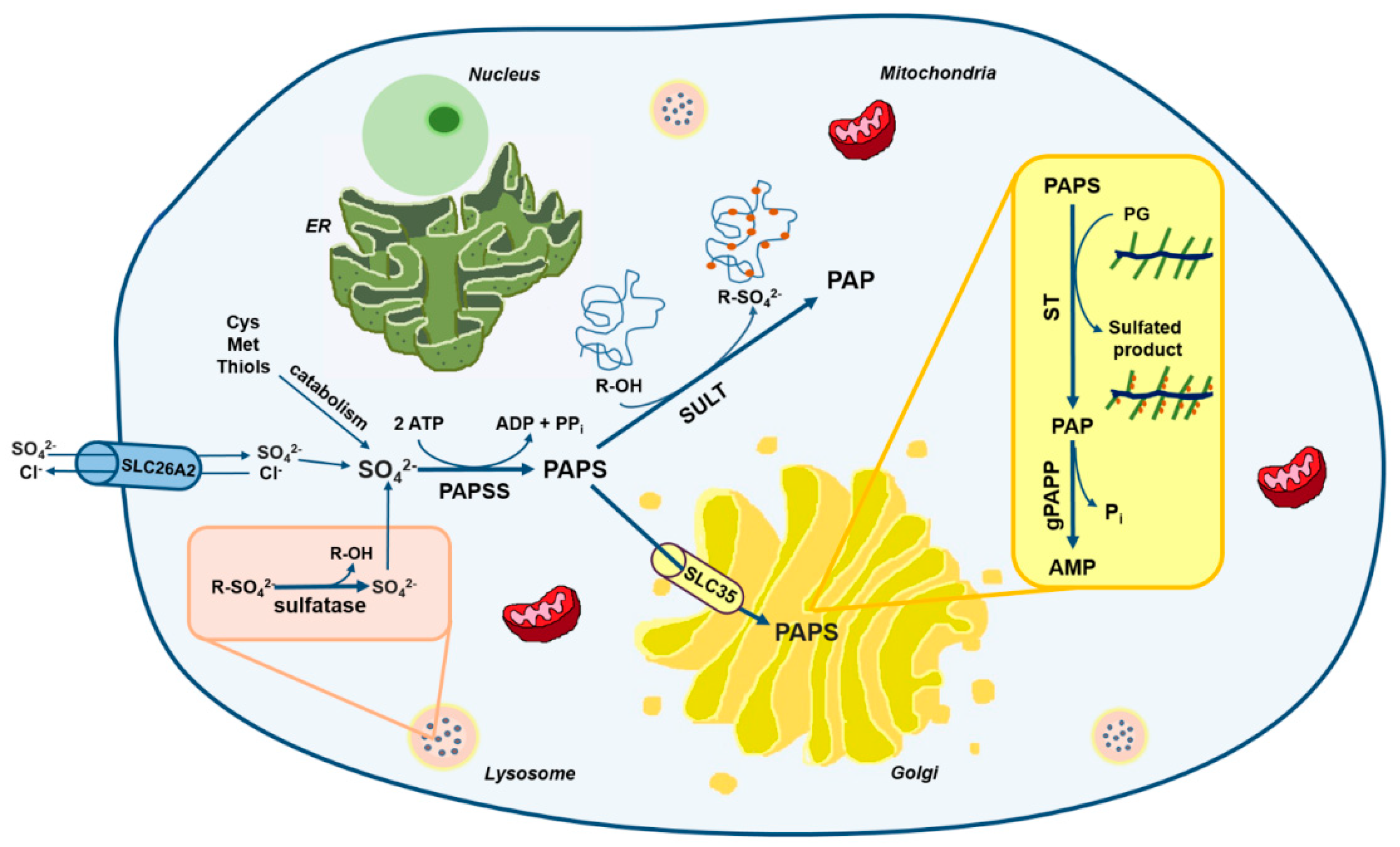
2. The Cellular Metabolism of Sulfate
2.1. The Origin of Intracellular Sulfate
2.2. The Sulfate Activation Pathway
2.3. The Sulfation Pathway
3. Sulfate Metabolism and Genetic Diseases
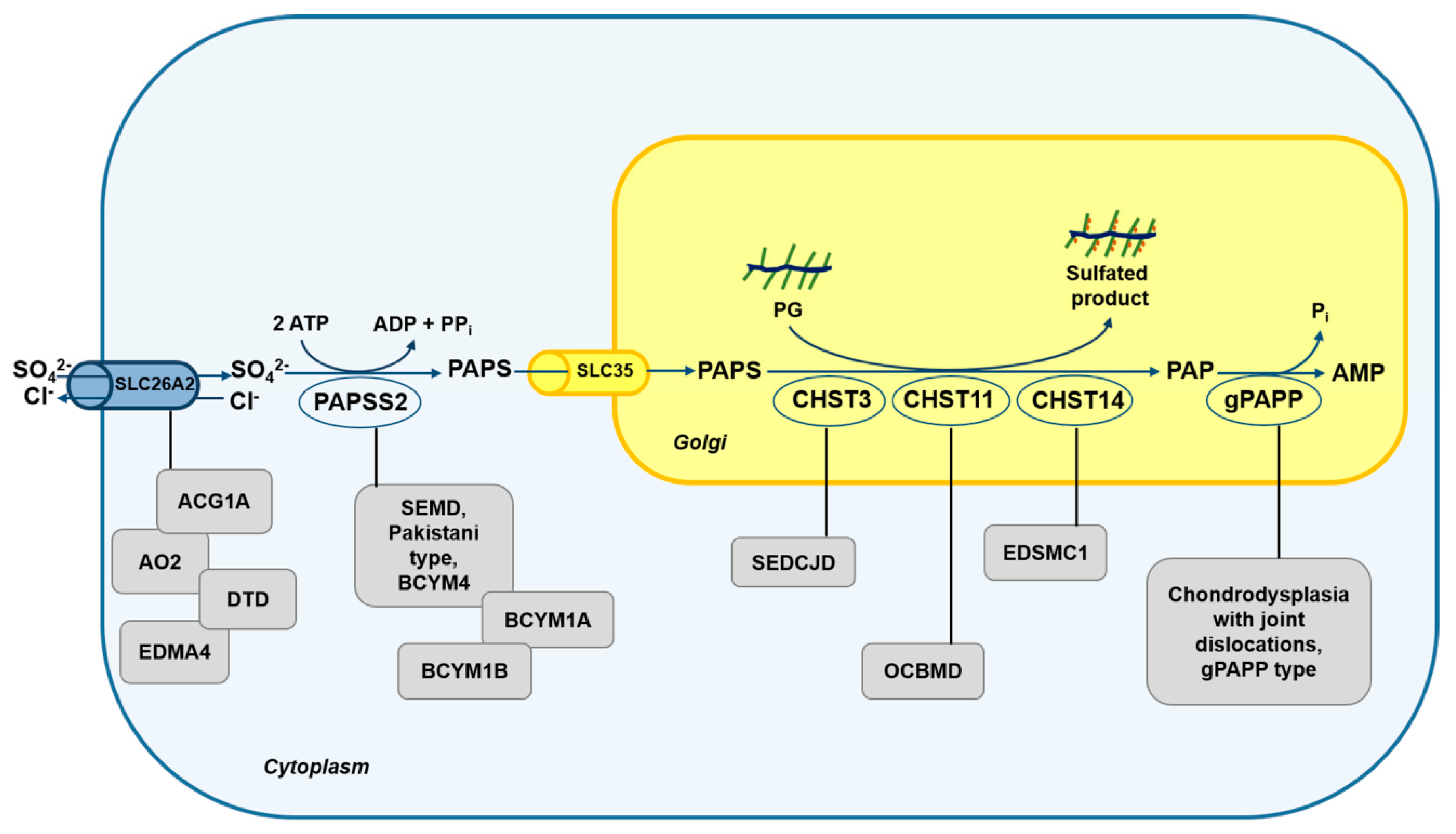
3.1. Skeletal Dysplasias Linked to Proteins Involved in Sulfate Metabolism
3.2. Skeletal Dysplasias Linked to Proteins Involved in GAG Sulfation
4. Alterations of Extracellular Matrix and Cell Homeostasis Due to Defects in PG Sulfation
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACG1B | achondrogenesis type 1B |
| AO2 | atelosteogenesis type 2 |
| APS | adenosine 5′-phosphosulfate |
| BCYM1A | brachyolmia type 1 Hobaek form |
| BCYM1B | brachyolmia type 1 Toledo form |
| BCYM4 | brachyolmia type 4 |
| BPNT2 | 3′(2′) 5′-bisphosphate nucleotidase 2 |
| C4ST | chondroitin-4-O-sulfotransferase |
| C6ST | chondroitin-6-O-sulfotransferase |
| CS | chondroitin sulfate |
| D4ST | dermatan-4-O-sulfotransferase |
| DHEA | dehydroepiandrosterone |
| DHEAS | dehydroepiandrosterone sulfate ester |
| DS | dermatan sulfate |
| DTD | diastrophic dysplasia |
| DTDST | diastrophic dysplasia sulfate transporter |
| ECM | extracellular matrix |
| EDM4 | recessive multiple epiphyseal dysplasia |
| EDSMC1 | Ehlers-Danlos syndrome musculocontractural type 1 |
| GAG | glycosaminoglycan |
| GalNAc | N-acetylgalactosamine |
| gPAPP | Golgi resident phosphoadenosine phosphate phosphatase |
| HS | heparan sulfate |
| IMPAD1 | inositol monophosphatase domain-containing protein 1 |
| OCBMD | osteochondrodysplasia brachydactyly and overlapping malformed digits |
| PAP | phosphoadenosine phosphate |
| PAPS | 3′-phosphoadenosine 5′-phosphosulfate |
| PAPSS | PAPS synthetase |
| PAPST | PAPS transporter |
| PG | proteoglycan |
| SEDCJD | spondyloepiphyseal dysplasia with congenital joint dislocations |
| ST | carbohydrate sulfotransferase |
| SULT | cytosolic sulfotransferase |
| TPST | tyrosylprotein sulfotransferase |
References
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [PubMed]
- Honke, K.; Taniguchi, N. Sulfotransferases and sulfated oligosaccharides. Med. Res. Rev. 2002, 22, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Bowman, K.G.; Bertozzi, C.R. Carbohydrate sulfotransferases: Mediators of extracellular communication. Chem. Biol. 1999, 6, R9–R22. [Google Scholar] [CrossRef]
- Gallagher, J.T. Heparan sulphates as membrane receptors for the fibroblast growth factors. Eur. J. Clin. Chem. Clin. Biochem. 1994, 32, 239–247. [Google Scholar]
- Pacifici, G.M. Sulfation of drugs and hormones in mid-gestation human fetus. Early Hum. Dev. 2005, 81, 573–581. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A. On the role of sulfolipids in mammalian metabolism. Mol. Cell. Biochem. 1985, 66, 87–95. [Google Scholar] [CrossRef]
- Stone, M.J.; Chuang, S.; Hou, X.; Shoham, M.; Zhu, J.Z. Tyrosine sulfation: An increasingly recognised post-translational modification of secreted proteins. Nat. Biotechnol. 2009, 25, 299–317. [Google Scholar] [CrossRef]
- Richard, K.; Hume, R.; Kaptein, E.; Stanley, E.L.; Visser, T.J.; Coughtrie, M.W. Sulfation of thyroid hormone and dopamine during human development: Ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J. Clin. Endocrinol. Metab. 2001, 86, 2734–2742. [Google Scholar] [CrossRef]
- Aksoy, I.A.; Otterness, D.M.; Weinshilboum, R.M. Cholesterol sulfation in human liver. Catalysis by dehydroepiandrosterone sulfotransferase. Drug Metab. Dispos. 1993, 21, 268–276. [Google Scholar]
- Alnouti, Y. Bile Acid sulfation: A pathway of bile acid elimination and detoxification. Toxicol. Sci. 2009, 108, 225–246. [Google Scholar] [CrossRef]
- Falany, C.N.; Wheeler, J.; Oh, T.S.; Falany, J.L. Steroid sulfation by expressed human cytosolic sulfotransferases. J. Steroid Biochem. Mol. Biol. 1994, 48, 369–375. [Google Scholar] [CrossRef]
- Paganini, C.; Costantini, R.; Superti-Furga, A.; Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: A panoramic view. FEBS J. 2019, 286, 3008–3032. [Google Scholar] [CrossRef]
- Krakow, D.; Rimoin, D.L. The skeletal dysplasias. Genet. Med. 2010, 12, 327–341. [Google Scholar] [CrossRef]
- Geister, K.A.; Camper, S.A. Advances in Skeletal Dysplasia Genetics. Annu. Rev. Genom. Hum. Genet. 2015, 16, 199–227. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Boles, J.W. Sulfation and sulfotransferases 5: The importance of 3’- phosphoadenosine 5’-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J. 1997, 11, 404–418. [Google Scholar] [CrossRef]
- Leyh, T.S. The physical biochemistry and molecular genetics of sulfate activation. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 515–542. [Google Scholar] [CrossRef]
- Lipmann, F. Biological sulfate activation and transfer. Science 1958, 128, 575–580. [Google Scholar] [CrossRef]
- Farooqui, A.A. 3’-phosphoadenosine 5’-phosphosulphate metabolism in mammalian tissues. Int. J. Biochem. 1980, 12, 529–536. [Google Scholar] [CrossRef]
- Venkatachalam, K.V. Human 3’-phosphoadenosine 5’-phosphosulfate (PAPS) synthase: Biochemistry, molecular biology and genetic deficiency. Iubmb Life 2003, 55, 1–11. [Google Scholar] [CrossRef]
- Langford, R.; Hurrion, E.; Dawson, P.A. Genetics and pathophysiology of mammalian sulfate biology. J. Genet. Genom. 2017, 44, 7–20. [Google Scholar] [CrossRef]
- Dawson, P.A. Sulfate in fetal development. Semin. Cell Dev. Biol. 2011, 22, 653–659. [Google Scholar] [CrossRef]
- Dawson, P.A.; Richard, K.; Perkins, A.; Zhang, Z.; Simmons, D.G. Review: Nutrient sulfate supply from mother to fetus: Placental adaptive responses during human and animal gestation. Placenta 2017, 54, 45–51. [Google Scholar] [CrossRef]
- Cole, D.E.; Scriver, C.R. Age-dependent serum sulfate levels in children and adolescents. Clin. Chim. Acta 1980, 107, 135–139. [Google Scholar] [CrossRef]
- Cole, D.E.; Baldwin, L.S.; Stirk, L.J. Increased serum sulfate in pregnancy: Relationship to gestational age. Clin. Chem. 1985, 31, 866–867. [Google Scholar] [CrossRef]
- Bradley, H.; Gough, A.; Sokhi, R.S.; Hassell, A.; Waring, R.; Emery, P. Sulfate metabolism is abnormal in patients with rheumatoid arthritis. Confirmation by in vivo biochemical findings. J. Rheumatol. 1994, 21, 1192–1196. [Google Scholar]
- Jennings, M.L. Proton fluxes associated with erythrocyte membrane anion exchange. J. Membr. Biol. 1976, 28, 187–205. [Google Scholar] [CrossRef]
- Lötscher, M.; Custer, M.; Quabius, E.S.; Kaissling, B.; Murer, H.; Biber, J. Immunolocalization of Na/SO4-cotransport (NaSi-1) in rat kidney. Pflug. Arch. 1996, 432, 373–378. [Google Scholar] [CrossRef]
- Simmons, D.G.; Rakoczy, J.; Jefferis, J.; Lourie, R.; McIntyre, H.D.; Dawson, P.A. Human placental sulfate transporter mRNA profiling from term pregnancies identifies abundant SLC13A4 in syncytiotrophoblasts and SLC26A2 in cytotrophoblasts. Placenta 2013, 34, 381–384. [Google Scholar] [CrossRef]
- Dawson, P.A.; Markovich, D. Pathogenetics of the human SLC26 transporters. Curr. Med. Chem. 2005, 12, 385–396. [Google Scholar] [CrossRef]
- Alper, S.L.; Sharma, A.K. The SLC26 gene family of anion transporters and channels. Mol. Asp. Med. 2013, 34, 494–515. [Google Scholar] [CrossRef]
- Markovich, D. Na+-sulfate cotransporter SLC13A1. Pflug. Arch. 2014, 466, 131–137. [Google Scholar] [CrossRef]
- Zhang, Z.; Aung, Z.T.; Simmons, D.G.; Dawson, P.A. Molecular analysis of sequence and splice variants of the human SLC13A4 sulfate transporter. Mol. Genet. Metab. 2017, 121, 35–42. [Google Scholar] [CrossRef]
- Hästbacka, J.; de la Chapelle, A.; Mahtani, M.M.; Clines, G.; Reeve Daly, M.P.; Daly, M.; Hamilton, B.A.; Kusumi, K.; Trivedi, B.; Weaver, A.; et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: Positional cloning by fine-structure linkage disequilibrium mapping. Cell 1994, 78, 1073–1087. [Google Scholar] [CrossRef]
- Dawson, J.R.; Norbeck, K.; Moldeus, P. The effectiveness of different sulfate precursors in supporting extrahepatic sulfate conjugation. Biochem. Pharm. 1983, 32, 1789–1791. [Google Scholar] [CrossRef]
- Elgavish, A.; Meezan, E. Sulfation by human lung fibroblasts: SO4(2-) and sulfur- containing amino acids as sources for macromolecular sulfation. Am. J. Physiol. 1991, 260, L450–L456. [Google Scholar] [CrossRef]
- Rome, L.H.; Hill, D.F. Lysosomal degradation of glycoproteins and glycosaminoglycans. Efflux and recycling of sulphate and N-acetylhexosamines. Biochem. J. 1986, 235, 707–713. [Google Scholar] [CrossRef]
- Lyle, S.; Stanczak, J.; Ng, K.; Schwartz, N.B. Rat chondrosarcoma ATP sulfurylase and adenosine 5’-phosphosulfate kinase reside on a single bifunctional protein. Biochemistry 1994, 33, 5920–5925. [Google Scholar] [CrossRef]
- Xu, Z.H.; Otterness, D.M.; Freimuth, R.R.; Carlini, E.J.; Wood, T.C.; Mitchell, S.; Moon, E.; Kim, U.J.; Xu, J.P.; Siciliano, M.J.; et al. Human 3’-phosphoadenosine 5’-phosphosulfate synthetase 1 (PAPSS1) and PAPSS2: Gene cloning, characterization and chromosomal localization. Biochem. Biophys. Res. Commun. 2000, 268, 437–444. [Google Scholar] [CrossRef]
- ul Haque, M.F.; King, L.M.; Krakow, D.; Cantor, R.M.; Rusiniak, M.E.; Swank, R.T.; Superti-Furga, A.; Haque, S.; Abbas, H.; Ahmad, W.; et al. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat. Genet. 1998, 20, 157–162. [Google Scholar] [CrossRef]
- Kurima, K.; Warman, M.L.; Krishnan, S.; Domowicz, M.; Krueger, R.C., Jr.; Deyrup, A.; Schwartz, N.B. A member of a family of sulfate-activating enzymes causes murine brachymorphism. Proc. Natl. Acad. Sci. USA 1998, 95, 8681–8685, Erratum in 1998, 95, 12071. [Google Scholar] [CrossRef] [PubMed]
- Noordam, C.; Dhir, V.; McNelis, J.C.; Schlereth, F.; Hanley, N.A.; Krone, N.; Smeitink, J.A.; Smeets, R.; Sweep, F.C.; Claahsen-van der Grinten, H.L.; et al. Inactivating PAPSS2 mutations in a patient with premature pubarche. N. Engl. J. Med. 2009, 360, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef] [PubMed]
- Fuda, H.; Shimizu, C.; Lee, Y.C.; Akita, H.; Strott, C.A. Characterization and expression of human bifunctional 3’-phosphoadenosine 5’-phosphosulphate synthase isoforms. Biochem. J. 2002, 365, 497–504. [Google Scholar] [CrossRef]
- Schroder, E.; Gebel, L.; Eremeev, A.A.; Morgner, J.; Grum, D.; Knauer, S.K.; Bayer, P.; Mueller, J.W. Human PAPS Synthase Isoforms Are Dynamically Regulated Enzymes with Access to Nucleus and Cytoplasm. PLoS ONE 2012, 7, e29559. [Google Scholar] [CrossRef]
- Kamiyama, S.; Sasaki, N.; Goda, E.; Ui-Tei, K.; Saigo, K.; Narimatsu, H.; Jigami, Y.; Kannagi, R.; Irimura, T.; Nishihara, S. Molecular cloning and characterization of a novel 3’-phosphoadenosine 5’-phosphosulfate transporter, PAPST2. J. Biol. Chem. 2006, 281, 10945–10953. [Google Scholar] [CrossRef]
- Kamiyama, S.; Suda, T.; Ueda, R.; Suzuki, M.; Okubo, R.; Kikuchi, N.; Chiba, Y.; Goto, S.; Toyoda, H.; Saigo, K.; et al. Molecular cloning and identification of 3’-phosphoadenosine 5’-phosphosulfate transporter. J. Biol. Chem. 2003, 278, 25958–25963. [Google Scholar] [CrossRef]
- Günal, S.; Hardman, R.; Kopriva, S.; Mueller, J.W. Sulfation pathways from red to green. J. Biol. Chem. 2019, 294, 12293–12312. [Google Scholar] [CrossRef]
- Parker, J.L.; Newstead, S. Gateway to the Golgi: Molecular mechanisms of nucleotide sugar transporters. Curr. Opin. Struct. Biol. 2019, 57, 127–134. [Google Scholar] [CrossRef]
- Wiweger, M.I.; Avramut, C.M.; de Andrea, C.E.; Prins, F.A.; Koster, A.J.; Ravelli, R.B.; Hogendoorn, P.C. Cartilage ultrastructure in proteoglycan-deficient zebrafish mutants brings to light new candidate genes for human skeletal disorders. J. Pathol. 2011, 223, 531–542. [Google Scholar] [CrossRef]
- Bojarova, P.; Williams, S.J. Sulfotransferases, sulfatases and formylglycine-generating enzymes: A sulfation fascination. Curr. Opin. Chem. Biol. 2008, 12, 573–581. [Google Scholar] [CrossRef]
- Coughtrie, M.W.H. Function and organization of the human cytosolic sulfotransferase (SULT) family. Chem. Biol. Interact. 2016, 259, 2–7. [Google Scholar] [CrossRef]
- Petrotchenko, E.V.; Pedersen, L.C.; Borchers, C.H.; Tomer, K.B.; Negishi, M. The dimerization motif of cytosolic sulfotransferases. FEBS Lett. 2001, 490, 39–43. [Google Scholar] [CrossRef]
- Hartmann-Fatu, C.; Trusch, F.; Moll, C.N.; Michin, I.; Hassinen, A.; Kellokumpu, S.; Bayer, P. Heterodimers of tyrosylprotein sulfotransferases suggest existence of a higher organization level of transferases in the membrane of the trans-Golgi apparatus. J. Mol. Biol. 2015, 427, 1404–1412. [Google Scholar] [CrossRef]
- Leung, A.W.; Backstrom, I.; Bally, M.B. Sulfonation, an underexploited area: From skeletal development to infectious diseases and cancer. Oncotarget 2016, 7, 55811–55827. [Google Scholar] [CrossRef]
- Vissers, L.E.; Lausch, E.; Unger, S.; Campos-Xavier, A.B.; Gilissen, C.; Rossi, A.; Del Rosario, M.; Venselaar, H.; Knoll, U.; Nampoothiri, S.; et al. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am. J. Hum. Genet. 2011, 88, 608–615. [Google Scholar] [CrossRef]
- Frederick, J.P.; Tafari, A.T.; Wu, S.M.; Megosh, L.C.; Chiou, S.T.; Irving, R.P.; York, J.D. A role for a lithium-inhibited Golgi nucleotidase in skeletal development and sulfation. Proc. Natl. Acad. Sci. USA 2008, 105, 11605–11612. [Google Scholar] [CrossRef]
- Sohaskey, M.L.; Yu, J.; Diaz, M.A.; Plaas, A.H.; Harland, R.M. JAWS coordinates chondrogenesis and synovial joint positioning. Development 2008, 135, 2215–2220. [Google Scholar] [CrossRef]
- Diez-Roux, G.; Ballabio, A. Sulfatases and human disease. Annu. Rev. Genom. Hum. Genet. 2005, 6, 355–379. [Google Scholar] [CrossRef]
- Rossi, A.; Superti-Furga, A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 Novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum. Mutat. 2001, 17, 159–171. [Google Scholar] [CrossRef]
- De la Chapelle, A.; Maroteaux, P.; Havu, N.; Granroth, G. A rare lethal bone dysplasia with recessive autosomic transmission. Arch. Fr. Pediatr. 1972, 29, 759–770. [Google Scholar]
- McAlister, W.H.; Crane, J.P.; Bucy, R.P.; Craig, R.B. A new neonatal short limbed dwarfism. Skelet. Radiol. 1985, 13, 271–275. [Google Scholar] [CrossRef]
- Bonafe, L.; Hastbacka, J.; de la Chapelle, A.; Campos-Xavier, A.B.; Chiesa, C.; Forlino, A.; Superti-Furga, A.; Rossi, A. A novel mutation in the sulfate transporter gene SLC26A2 (DTDST) specific to the Finnish population causes de la Chapelle dysplasia. J. Med. Genet. 2008, 45, 827–831. [Google Scholar] [CrossRef]
- Rossi, A.; Bonaventure, J.; Delezoide, A.L.; SupertiFurga, A.; Cetta, G. Undersulfation of cartilage proteoglycans ex vivo and increased contribution of amino acid sulfur to sulfation in vitro in McAlister dysplasia atelosteogenesis type 2. Eur. J. Biochem. 1997, 248, 741–747. [Google Scholar] [CrossRef]
- Karniski, L.P. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene: Correlation between sulfate transport activity and chondrodysplasia phenotype. Hum. Mol. Genet. 2001, 10, 1485–1490. [Google Scholar] [CrossRef]
- Rossi, A.; Kaitila, I.; Wilcox, W.R.; Rimoin, D.L.; Steinmann, B.; Cetta, G.; Superti-Furga, A. Proteoglycan sulfation in cartilage and cell cultures from patients with sulfate transporter chondrodysplasias: Relationship to clinical severity and indications on the role of intracellular sulfate production. Matrix Biol. 1998, 17, 361–369. [Google Scholar] [CrossRef]
- Forlino, A.; Piazza, R.; Tiveron, C.; Della Torre, S.; Tatangelo, L.; Bonafe, L.; Gualeni, B.; Romano, A.; Pecora, F.; Superti-Furga, A.; et al. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: Morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum. Mol. Genet. 2005, 14, 859–871. [Google Scholar] [CrossRef]
- Gualeni, B.; Facchini, M.; De Leonardis, F.; Tenni, R.; Cetta, G.; Viola, M.; Passi, A.; Superti-Furga, A.; Forlino, A.; Rossi, A. Defective proteoglycan sulfation of the growth plate zones causes reduced chondrocyte proliferation via an altered Indian hedgehog signalling. Matrix Biol. 2010, 29, 453–460. [Google Scholar] [CrossRef]
- De Leonardis, F.; Monti, L.; Gualeni, B.; Tenni, R.; Forlino, A.; Rossi, A. Altered signaling in the G1 phase deregulates chondrocyte growth in a mouse model with proteoglycan undersulfation. J. Cell. Biochem. 2014, 115, 1779–1786. [Google Scholar] [CrossRef]
- Monti, L.; Paganini, C.; Lecci, S.; De Leonardis, F.; Hay, E.; Cohen-Solal, M.; Villani, S.; Superti-Furga, A.; Tenni, R.; Forlino, A.; et al. N-acetylcysteine treatment ameliorates the skeletal phenotype of a mouse model of diastrophic dysplasia. Hum. Mol. Genet. 2015, 24, 5570–5580. [Google Scholar] [CrossRef]
- Zheng, C.; Lin, X.; Xu, X.; Wang, C.; Zhou, J.; Gao, B.; Fan, J.; Lu, W.; Hu, Y.; Jie, Q.; et al. Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias. EBioMedicine 2019, 40, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Haque, M.F.; Ahmad, W.; Abbas, H.; Haque, S.; Krakow, D.; Rimoin, D.L.; Lachman, R.S.; Cohn, D.H. Distinct, autosomal recessive form of spondyloepimetaphyseal dysplasia segregating in an inbred Pakistani kindred. Am. J. Med. Genet. 1998, 78, 468–473. [Google Scholar] [CrossRef]
- Miyake, N.; Elcioglu, N.H.; Iida, A.; Isguven, P.; Dai, J.; Murakami, N.; Takamura, K.; Cho, T.J.; Kim, O.H.; Hasegawa, T.; et al. PAPSS2 mutations cause autosomal recessive brachyolmia. J. Med. Genet. 2012, 49, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Toledo, S.P.; Mourao, P.A.; Lamego, C.; Alves, C.A.; Dietrich, C.P.; Assis, L.M.; Mattar, E. Recessively inherited, late onset spondylar dysplasia and peripheral corneal opacity with anomalies in urinary mucopolysaccharides: A possible error of chondroitin-6-sulfate synthesis. Am. J. Med. Genet. 1978, 2, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Mourao, P.A.; Kato, S.; Donnelly, P.V. Spondyloepiphyseal dysplasia, chondroitin sulfate type: A possible defect of PAPS--chondroitin sulfate sulfotransferase in humans. Biochem. Biophys. Res. Commun. 1981, 98, 388–396. [Google Scholar] [CrossRef]
- Iida, A.; Simsek-Kiper, P.O.; Mizumoto, S.; Hoshino, T.; Elcioglu, N.; Horemuzova, E.; Geiberger, S.; Yesil, G.; Kayserili, H.; Utine, G.E.; et al. Clinical and radiographic features of the autosomal recessive form of brachyolmia caused by PAPSS2 mutations. Hum. Mutat. 2013, 34, 1381–1386. [Google Scholar] [CrossRef]
- Nizon, M.; Alanay, Y.; Tuysuz, B.; Kiper, P.O.; Genevieve, D.; Sillence, D.; Huber, C.; Munnich, A.; Cormier-Daire, V. IMPAD1 mutations in two Catel-Manzke like patients. Am. J. Med. Genet. A 2012, 158A, 2183–2187. [Google Scholar] [CrossRef]
- Unger, S.; Lausch, E.; Rossi, A.; Megarbane, A.; Sillence, D.; Alcausin, M.; Aytes, A.; Mendoza-Londono, R.; Nampoothiri, S.; Afroze, B.; et al. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: Congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. A 2010, 152A, 2543–2549. [Google Scholar] [CrossRef]
- Thiele, H.; Sakano, M.; Kitagawa, H.; Sugahara, K.; Rajab, A.; Hohne, W.; Ritter, H.; Leschik, G.; Nurnberg, P.; Mundlos, S. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA 2004, 101, 10155–10160. [Google Scholar] [CrossRef]
- Hermanns, P.; Unger, S.; Rossi, A.; Perez-Aytes, A.; Cortina, H.; Bonafe, L.; Boccone, L.; Setzu, V.; Dutoit, M.; Sangiorgi, L.; et al. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 2008, 82, 1368–1374. [Google Scholar] [CrossRef]
- van Roij, M.H.; Mizumoto, S.; Yamada, S.; Morgan, T.; Tan-Sindhunata, M.B.; Meijers-Heijboer, H.; Verbeke, J.I.; Markie, D.; Sugahara, K.; Robertson, S.P. Spondyloepiphyseal dysplasia, Omani type: Further definition of the phenotype. Am. J. Med. Genet. A 2008, 146A, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Tuysuz, B.; Mizumoto, S.; Sugahara, K.; Celebi, A.; Mundlos, S.; Turkmen, S. Omani-type spondyloepiphyseal dysplasia with cardiac involvement caused by a missense mutation in CHST3. Clin. Genet. 2009, 75, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, K.S.; Celermajer, J.M.; Tink, A.R. Humero-spinal dysostosis with congenital heart disease. Am. J. Dis. Child. 1974, 127, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.D. Humero-spinal dysostosis: Report of the fourth case with emphasis on generalized skeletal involvement, abnormal craniofacial features, and mitral valve thickening. J. Pediatr. Orthop. B 1997, 6, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Le, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Muller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170, 103–115. [Google Scholar] [CrossRef]
- Dundar, M.; Muller, T.; Zhang, Q.; Pan, J.; Steinmann, B.; Vodopiutz, J.; Gruber, R.; Sonoda, T.; Krabichler, B.; Utermann, G.; et al. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009, 85, 873–882. [Google Scholar] [CrossRef]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef]
- Hirose, T.; Takahashi, N.; Tangkawattana, P.; Minaguchi, J.; Mizumoto, S.; Yamada, S.; Miyake, N.; Hayashi, S.; Hatamochi, A.; Nakayama, J.; et al. Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta Gen. Subj. 2018. [Google Scholar] [CrossRef]
- Shabbir, R.M.K.; Nalbant, G.; Ahmad, N.; Malik, S.; Tolun, A. Homozygous CHST11 mutation in chondrodysplasia, brachydactyly, overriding digits, clino-symphalangism and synpolydactyly. J. Med. Genet. 2018, 55, 489–496. [Google Scholar] [CrossRef]
- Chopra, S.S.; Leshchiner, I.; Duzkale, H.; McLaughlin, H.; Giovanni, M.; Zhang, C.; Stitziel, N.; Fingeroth, J.; Joyce, R.M.; Lebo, M.; et al. Inherited CHST11/MIR3922 deletion is associated with a novel recessive syndrome presenting with skeletal malformation and malignant lymphoproliferative disease. Mol. Genet. Genom. Med. 2015, 3, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Kluppel, M.; Wight, T.N.; Chan, C.; Hinek, A.; Wrana, J.L. Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development 2005, 132, 3989–4003. [Google Scholar] [CrossRef] [PubMed]
- SupertiFurga, A.; Hastbacka, J.; Wilcox, W.R.; Cohn, D.H.; vanderHarten, H.J.; Rossi, A.; Blau, N.; Rimoin, D.L.; Steinmann, B.; Lander, E.S.; et al. Achondrogenesis type IB is caused by mutations in the diastrophic dysplasia sulphate transporter gene. Nat. Genet. 1996, 12, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Hästbacka, J.; Superti-Furga, A.; Wilcox, W.R.; Rimoin, D.L.; Cohn, D.H.; Lander, E.S. Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): Evidence for a phenotypic series involving three chondrodysplasias. Am. J. Hum. Genet. 1996, 58, 255–262. [Google Scholar]
- Superti-Furga, A.; Neumann, L.; Riebel, T.; Eich, G.; Steinmann, B.; Spranger, J.; Kunze, J. Recessively inherited multiple epiphyseal dysplasia with normal stature, club foot, and double layered patella caused by a DTDST mutation. J. Med. Genet. 1999, 36, 621–624. [Google Scholar]
- Gualeni, B.; de Vernejoul, M.C.; Marty-Morieux, C.; De Leonardis, F.; Franchi, M.; Monti, L.; Forlino, A.; Houillier, P.; Rossi, A.; Geoffroy, V. Alteration of proteoglycan sulfation affects bone growth and remodeling. Bone 2013, 54, 83–91. [Google Scholar] [CrossRef]
- Cortes, M.; Baria, A.T.; Schwartz, N.B. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development 2009, 136, 1697–1706. [Google Scholar] [CrossRef]
- Besio, R.; Antonella, F. Treatment options for osteogenesis imperfecta. Expert Opin. Orphan Drugs 2015, 3, 165–181. [Google Scholar] [CrossRef]
- Briggs, M.D.; Bell, P.A.; Wright, M.J.; Pirog, K.A. New therapeutic targets in rare genetic skeletal diseases. Expert Opin. Orphan Drugs 2015, 3, 1137–1154. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Legeai-Mallet, L. Achondroplasia: Development, pathogenesis, and therapy. Dev. Dyn. 2017, 246, 291–309. [Google Scholar] [CrossRef]
- Marzin, P.; Cormier-Daire, V. New perspectives on the treatment of skeletal dysplasia. Adv. Endocrinol. Metab. 2020, 11. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Pathology | MIM/Inheritance | Causative Gene | Protein Product and Function | Biochemical Phenotype | References |
|---|---|---|---|---|---|
| Achondrogenesis type 1B (ACG1B) | 600972/AR | SLC26A2 | Sulfate/chloride antiporter present on cell membrane. | Severe cartilage PG undersulfation; reduced sulfate uptake in fibroblasts. | [60,65,66,71,93] |
| Atelosteogenesis type 2 (AO2) | 256050/AR | SLC26A2 | Sulfate/chloride antiporter present on cell membrane. | Severe cartilage PG undersulfation; reduced sulfate uptake in fibroblasts. | [60,65,66,94] |
| Diastrophic dysplasia (DTD) | 222600/AR | SLC26A2 | Sulfate/chloride antiporter present on cell membrane. | Cartilage PG undersulfation; reduced sulfate uptake in fibroblasts; in mice altered sulfate uptake in chondrocytes, and osteoblasts; altered Ihh signaling; reduced chondrocytes proliferation. | [34,60,65,66,67,68,69,70] |
| Recessive multiple epiphyseal dysplasia (EDM4) | 226900/AR | SLC26A2 | Sulfate/chloride antiporter present on cell membrane. | Reduced sulfate uptake. | [60,65,95] |
| Spondyloepimetaphyseal dysplasia, SEMD, Pakistani type or Brachyolmia type 4 (BCYM4) | 612847/AR | PAPSS2 | PAPS synthetase 2, enzyme that synthesizes the universal sulfate donor (PAPS). | Macromolecular undersulfation; signs of androgen excess (in a minority of patients); very low DHEAS levels and increased androgen levels (in one patient). | [40,42,72,73,76] |
| Brachyolmia type 1 (includes Hobaek form and Toledo form, BCYM1A and 1B respectively) | 271530/AR 271630/AR | PAPSS2 | PAPS synthetase 2, enzyme that synthesizes the universal sulfate donor (PAPS). | Undersulfation of CS; low activity of PAPS-CS sulfotransferase; signs of androgen excess (in a minority of patients). | [73,74,75,76] |
| Chondrodysplasia with joint dislocations, gPAPP type | 614078/AR | BPNT2 | Golgi resident PAP phosphatase, enzyme that hydrolyzes PAP to AMP and phosphate. | In mice impaired CS and HS sulfation. | [56,57,58,77] |
| Spondyloepiphyseal dysplasia with congenital joint dislocations (SEDCJD or SED Omani type) | 143095/AR | CHST3 | Carbohydrate sulfotransferase-3, enzyme that transfers sulfate to GalNAc residues of CS. | Depletion of 6-O-sulfated GalNAc residues in CS chains in fibroblasts and urine. | [78,79,80,81,82,83,84] |
| Ehlers-Danlos syndrome musculocontractural type 1 (EDSMC1) | 601776/AR | CHST14 | Carbohydrate sulfotransferase-14, enzyme that transfers sulfate to GalNAc residues of DS. | Reduction of 4-O-sulfation in GalNAc residues in DS chains; decrease of DS and increase of CS chain synthesis. | [85,86,87,88,89] |
| Osteochondrodysplasia, brachydactyly and overlapping malformed digits (OCBMD) | 618167/AR | CHST11 | Carbohydrate sulfotransferase-11, enzyme that transfers sulfate to GalNAc residues of CS. | In mice abnormal CS localization; strong up-regulation of TGF-β signaling and down-regulation of BMP signaling; altered morphology of the growth plate. | [90,91,92] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paganini, C.; Gramegna Tota, C.; Superti-Furga, A.; Rossi, A. Skeletal Dysplasias Caused by Sulfation Defects. Int. J. Mol. Sci. 2020, 21, 2710. https://doi.org/10.3390/ijms21082710
Paganini C, Gramegna Tota C, Superti-Furga A, Rossi A. Skeletal Dysplasias Caused by Sulfation Defects. International Journal of Molecular Sciences. 2020; 21(8):2710. https://doi.org/10.3390/ijms21082710
Chicago/Turabian StylePaganini, Chiara, Chiara Gramegna Tota, Andrea Superti-Furga, and Antonio Rossi. 2020. "Skeletal Dysplasias Caused by Sulfation Defects" International Journal of Molecular Sciences 21, no. 8: 2710. https://doi.org/10.3390/ijms21082710
APA StylePaganini, C., Gramegna Tota, C., Superti-Furga, A., & Rossi, A. (2020). Skeletal Dysplasias Caused by Sulfation Defects. International Journal of Molecular Sciences, 21(8), 2710. https://doi.org/10.3390/ijms21082710