Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review

 ,
, 
 ,
, 
Abstract
1. Introduction
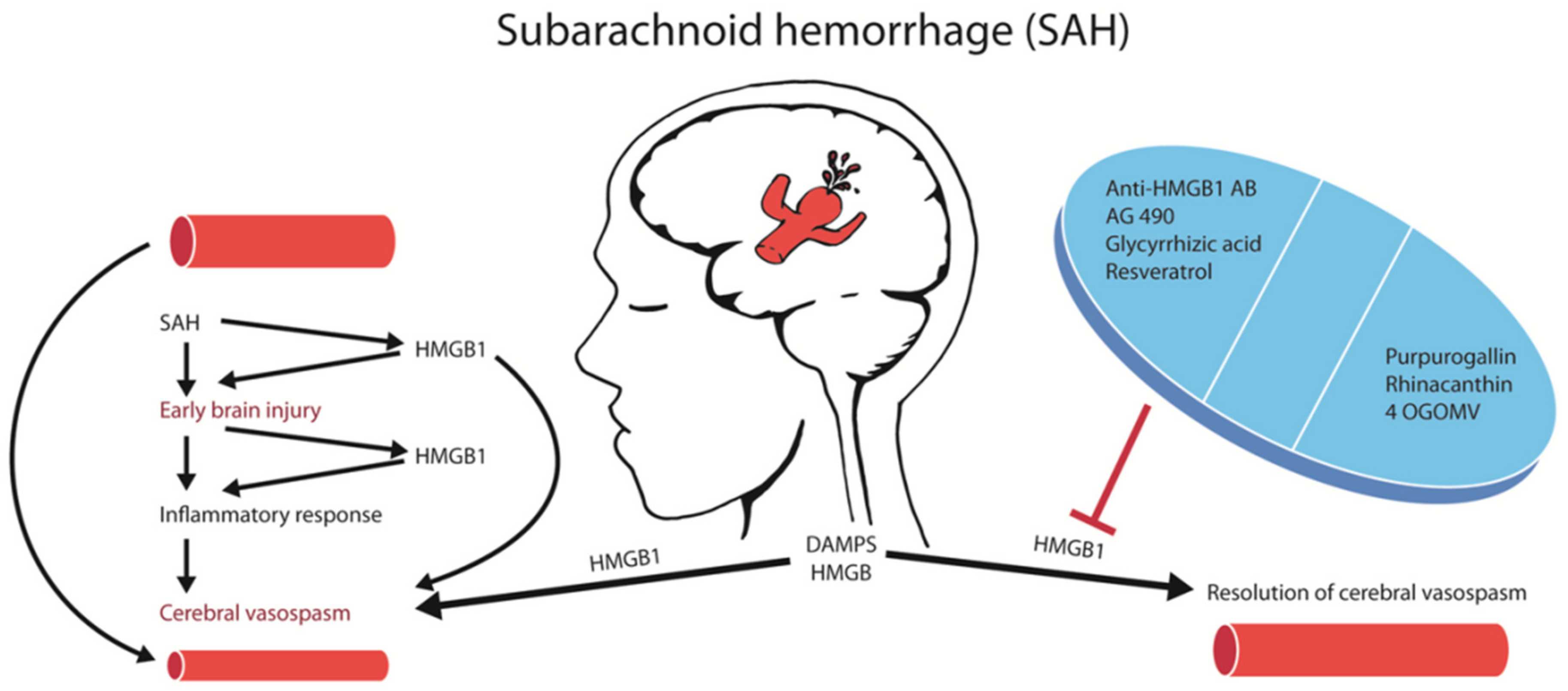
2. Results
2.1. HMGB1 Is Released in Cerebrospinal Fluid (CSF) and Systemic Circulation after aSAH
2.2. Pharmacological Inhibition of HMGB1 Protects against Early Brain Injury after SAH
2.3. Anti-HMGB1 Antibodies Confer Protection against CVS
2.4. Subarachnoid Hemorrhage and Blood–Brain Barrier
2.5. Soluble RAGE Protected against EBI
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grasso, G.; Alafaci, C.; Macdonald, R.L. Management of aneurysmal subarachnoid hemorrhage: State of the art and future perspectives. Surg. Neurol. Int. 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2013, 10, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Van Gijn, J.; Kerr, R.S.; Rinkel, G.J. Subarachnoid haemorrhage. Lancet 2007, 369, 306–318. [Google Scholar] [CrossRef]
- Etminan, N.; Rinkel, G.J. Unruptured intracranial aneurysms: Development, rupture and preventive management. Nat. Rev. Neurol. 2016, 12, 699–713. [Google Scholar] [CrossRef]
- Cahill, J.; Zhang, F. Subarachnoid hemorrhage: Is it time for a new direction? Stroke 2008, 40, S86–S87. [Google Scholar] [CrossRef]
- Suárez, J.I.; Tarr, R.W.; Selman, W.R. Aneurysmal Subarachnoid Hemorrhage. New Engl. J. Med. 2006, 354, 387–396. [Google Scholar] [CrossRef]
- Macdonald, R.L.; A Schweizer, T. Spontaneous subarachnoid haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Cahill, J.; Calvert, J.W.; Solaroglu, I.; Zhang, F. Vasospasm and p53-Induced Apoptosis in an Experimental Model of Subarachnoid Hemorrhage. Stroke 2006, 37, 1868–1874. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Hafez, A.; Jahromi, B.R.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. [Google Scholar] [CrossRef]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Pilzweger, C.; Holdenrieder, S. Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases. Diagn. 2015, 5, 219–253. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson-Harris, H.; Raucci, A. Alarmin(g) news about danger. EMBO Rep. 2006, 7, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a Late Mediator of Endotoxin Lethality in Mice. Sci. 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Tsuruta, R.; Kaneko, T.; Yamashita, S.; Fujita, M.; Kasaoka, S.; Hashiguchi, T.; Suzuki, M.; Maruyama, I.; Maekawa, T. High-Mobility Group Box 1 Protein in CSF of Patients with Subarachnoid Hemorrhage. Neurocritical Care 2009, 11, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; A Manfredi, A. IMMUNOLOGY: Dangers In and Out. Sci. 2009, 323, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Laird, M.D.; Ramesh, S.S.; Youssef, P.; Shakir, B.; Vender, J.R.; Alleyne, C.H.; Dhandapani, K.M.; Sangeetha, S. Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: An emerging role for neuroproteomics. Neurosurg. Focus 2010, 28, E10. [Google Scholar] [CrossRef]
- Murakami, K.; Koide, M.; Dumont, T.M.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Subarachnoid Hemorrhage Induces Gliosis and Increased Expression of the Pro-inflammatory Cytokine High Mobility Group Box 1 Protein. Transl. Stroke Res. 2010, 2, 72–79. [Google Scholar] [CrossRef]
- Zhu, X.-D.; Chen, J.-S.; Zhou, F.; Liu, Q.-C.; Chen, G.; Zhang, J.-M. Relationship between plasma high mobility group box-1 protein levels and clinical outcomes of aneurysmal subarachnoid hemorrhage. J. Neuroinflammation 2012, 9, 194. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Güresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Vatter, H.; Güresir, E.; Muhammad, S. Systemic High-Mobility Group Box-1. Crit. Care Med. 2018, 46, e1023–e1028. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Kahlert, U.D.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Hänggi, D.; Muhammad, S. Elevated Systemic IL-10 Levels Indicate Immunodepression Leading to Nosocomial Infections after Aneurysmal Subarachnoid Hemorrhage (SAH) in Patients. Int. J. Mol. Sci. 2020, 21, 1569. [Google Scholar] [CrossRef] [PubMed]
- Sokół, B.; Woźniak, A.; Jankowski, R.; Jurga, S.; Wąsik, N.; Shahid, H.; Grześkowiak, B.F. HMGB1 Level in Cerebrospinal Fluid as a Marker of Treatment Outcome in Patients with Acute Hydrocephalus Following Aneurysmal Subarachnoid Hemorrhage. J. Stroke Cerebrovasc. Dis. 2015, 24, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Francoeur, C.L.; Mayer, S.A. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit. Care 2016, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.D.; Rhind, S.G.; Di Battista, A.P.; Macdonald, R.L.; Baker, A.J. Biomarkers of Glycocalyx Injury are Associated with Delayed Cerebral Ischemia Following Aneurysmal Subarachnoid Hemorrhage: A Case Series Supporting a New Hypothesis. Neurocritical Care 2016, 26, 339–347. [Google Scholar] [CrossRef]
- Hendrix, P.; Foreman, P.; Harrigan, M.R.; Fisher, W.S.; Vyas, N.A.; Lipsky, R.; Lin, M.; Walters, B.; Tubbs, R.S.; Shoja, M.M.; et al. Impact of High-Mobility Group Box 1 Polymorphism on Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. World Neurosurg. 2017, 101, 325–330. [Google Scholar] [CrossRef]
- Sun, Q.; Wu, W.; Hu, Y.-C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.-D.; Ma, B.; Zhu, J.-H.; et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflammation 2014, 11, 106. [Google Scholar] [CrossRef]
- Chang, C.-Z.; Lin, C.-L.; Wu, S.-C.; Kwan, A.-L. Purpurogallin, a Natural Phenol, Attenuates High-Mobility Group Box 1 in Subarachnoid Hemorrhage Induced Vasospasm in a Rat Model. Int. J. Vasc. Med. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Chang, C.-Z.; Wu, S.-C.; Kwan, A.-L.; Lin, C.-L. 4′-O-β-D-glucosyl-5-O-methylvisamminol, an active ingredient of Saposhnikovia divaricata, attenuates high-mobility group box 1 and subarachnoid hemorrhage-induced vasospasm in a rat model. Behav. Brain Funct. 2015, 11, 28. [Google Scholar] [CrossRef]
- Chang, C.-Z.; Wu, S.-C.; Kwan, A.-L.; Lin, C.-L.; Information, P.E.K.F.C. Rhinacanthin-C, A Fat-Soluble Extract from Rhinacanthus nasutus, Modulates High-Mobility Group Box 1-Related Neuro-Inflammation and Subarachnoid Hemorrhage-Induced Brain Apoptosis in a Rat Model. World Neurosurg. 2016, 86, 349–360. [Google Scholar] [CrossRef]
- Zhang, X.-S.; Li, W.; Wu, Q.; Wu, L.-Y.; Ye, Z.-N.; Liu, J.-P.; Zhuang, Z.; Zhou, M.-L.; Zhang, X.; Hang, C.-H. Resveratrol Attenuates Acute Inflammatory Injury in Experimental Subarachnoid Hemorrhage in Rats via Inhibition of TLR4 Pathway. Int. J. Mol. Sci. 2016, 17, 1331. [Google Scholar] [CrossRef]
- Zhao, X.D.; Mao, H.Y.; Lv, J.; Lu, X.J. Expression of high-mobility group box-1 (HMGB1) in the basilar artery after experimental subarachnoid hemorrhage. J. Clin. Neurosci. 2016, 27, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, F.; Jing, Z.; Wang, X.; Hua, X.; Wan, L. Glycyrrhizic acid exerts anti-inflammatory effect to improve cerebral vasospasm secondary to subarachnoid hemorrhage in a rat model. Neurol. Res. 2017, 39, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Ieong, C.; Sun, H.; Wang, Q.; Ma, J. Glycyrrhizin suppresses the expressions of HMGB1 and ameliorates inflammative effect after acute subarachnoid hemorrhage in rat model. J. Clin. Neurosci. 2018, 47, 278–284. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Pang, H.; Huang, T.; Song, J.; Li, D.; Zhao, Y.; Ma, X. AG490 ameliorates early brain injury via inhibition of JAK2/STAT3-mediated regulation of HMGB1 in subarachnoid hemorrhage. Exp. Ther. Med. 2017, 15, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Pan, H.; Li, J.; Xu, H.; Jin, H.; Qian, C.; Yan, F.; Chen, J.; Wang, C.; Chen, J.; et al. Inhibiting of RIPK3 attenuates early brain injury following subarachnoid hemorrhage: Possibly through alleviating necroptosis. Biomed. Pharmacother. 2018, 107, 563–570. [Google Scholar] [CrossRef]
- Xiong, L.; Sun, L.; Zhang, Y.; Peng, J.; Yan, J.; Liu, X.; Jin, P. Exosomes from Bone Marrow Mesenchymal Stem Cells Can Alleviate Early Brain Injury After Subarachnoid Hemorrhage Through miRNA129-5p-HMGB1 Pathway. Stem Cells Dev. 2020, 29, 212–221. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Buller, B.; Chopp, M. Exosomes—Beyond stem cells for restorative therapy in stroke and neurological injury. Nat. Rev. Neurol. 2019, 15, 193–203. [Google Scholar] [CrossRef]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Liang, L.; Wu, Y.; Zhong, J.; Sun, X. Anti-high mobility group box-1 antibody attenuated vascular smooth muscle cell phenotypic switching and vascular remodelling after subarachnoid haemorrhage in rats. Neurosci. Lett. 2019, 708, 134338. [Google Scholar] [CrossRef]
- O’Brown, N.M.; Pfau, S.J.; Gu, C. Bridging barriers: A comparative look at the blood–brain barrier across organisms. Genes Dev. 2018, 32, 466–478. [Google Scholar] [CrossRef]
- Suzuki, H.; Kanamaru, H. Potential therapeutic molecular targets for blood-brain barrier disruption after subarachnoid hemorrhage. Neural Regen. Res. 2019, 14, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Nishibori, M.; Mori, S.; Takahashi, H.K. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharmacol. Sci. 2019, 140, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood–brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef] [PubMed]
- Lublinsky, S.; Major, S.; Kola, V.; Horst, V.; Santos, E.; Platz, J.; Sakowitz, O.; Scheel, M.; Dohmen, C.; Graf, R.; et al. Early blood-brain barrier dysfunction predicts neurological outcome following aneurysmal subarachnoid hemorrhage. EBioMedicine 2019, 43, 460–472. [Google Scholar] [CrossRef]
- Ha, C.H.; Kim, S.; Chung, J.; An, S.H.; Park, S.; Choi, N.; Kwon, K. Inhibitory effect of soluble RAGE in disturbed flow-induced atherogenesis. Int. J. Mol. Med. 2013, 32, 373–380. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Li, H.; Wu, W.; Sun, Q.; Liu, M.; Li, W.; Zhang, X.; Zhou, M.-L.; Hang, C.-H. Expression and cell distribution of receptor for advanced glycation end-products in the rat cortex following experimental subarachnoid hemorrhage. Brain Res. 2014, 1543, 315–323. [Google Scholar] [CrossRef]
- Wang, K.; Tang, S.-C.; Lee, J.-E.; Li, Y.-I.; Huang, Y.-S.; Yang, W.-S.; Jeng, J.-S.; Arumugam, T.V.; Tu, Y.-K. Cerebrospinal fluid high mobility group box 1 is associated with neuronal death in subarachnoid hemorrhage. Br. J. Pharmacol. 2016, 37, 435–443. [Google Scholar] [CrossRef]
- Eisenhut, M. Vasospasm in Cerebral Inflammation. Int. J. Inflamm. 2014, 2014, 1–14. [Google Scholar] [CrossRef]
- Li, H.; Yu, J.-S.; Zhang, D.-D.; Yang, Y.-Q.; Huang, L.-T.; Yu, Z.; Chen, R.-D.; Yang, H.-K.; Hang, C.-H. Inhibition of the Receptor for Advanced Glycation End-Products (RAGE) Attenuates Neuroinflammation While Sensitizing Cortical Neurons Towards Death in Experimental Subarachnoid Hemorrhage. Mol. Neurobiol. 2016, 54, 755–767. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Yue, L.; Liu, H.; Yang, X.; Wang, X.; Lin, Y.; Qu, Y. Evidence for the protective effects of curcumin against oxyhemoglobin-induced injury in rat cortical neurons. Brain Res. Bull. 2016, 120, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Dong, Y.; Huo, R.; Wen, K.; Zhang, Y.; Liang, G. Rutin Inhibits Neuroinflammation and Provides Neuroprotection in an Experimental Rat Model of Subarachnoid Hemorrhage, Possibly Through Suppressing the RAGE–NF-κB Inflammatory Signaling Pathway. Neurochem. Res. 2016, 41, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-H.; Xiong, L.-L.; Yang, S.-F.; You, C.; Xia, Q.-J.; Xu, Y.; Zhang, P.; Wang, S.-F.; Liu, J. LPS Pretreatment Provides Neuroprotective Roles in Rats with Subarachnoid Hemorrhage by Downregulating MMP9 and Caspase3 Associated with TLR4 Signaling Activation. Mol. Neurobiol. 2016, 54, 7746–7760. [Google Scholar] [CrossRef]
- Sarrafzadeh, A.S.; Schlenk, F.; Meisel, A.; Dreier, J.P.; Vajkoczy, P.; Meisel, C. Immunodepression After Aneurysmal Subarachnoid Hemorrhage. Stroke 2011, 42, 53–58. [Google Scholar] [CrossRef]
- Tsubota, M.; Fukuda, R.; Hayashi, Y.; Miyazaki, T.; Ueda, S.; Yamashita, R.; Koike, N.; Sekiguchi, F.; Wake, H.; Wakatsuki, S.; et al. Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: Negative impact of anticoagulants. J. Neuroinflammation 2019, 16, 199. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Sun, L.; Feng, D.; Sun, Q.; Dou, Y.; Liu, C.; Zhou, F.; Li, H.; Shen, H.; Wang, Z.; et al. HMGB1 promotes neurovascular remodeling via Rage in the late phase of subarachnoid hemorrhage. Brain Res. 2017, 1670, 135–145. [Google Scholar] [CrossRef]
- Isshiki, T.; Sakamoto, S.; Homma, S. Therapeutic Role of Recombinant Human Soluble Thrombomodulin for Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Med. 2019, 55, 172. [Google Scholar] [CrossRef]
- Friedrich, B.; Müller, F.; Feiler, S.; Schöller, K.; Plesnila, N. Experimental Subarachnoid Hemorrhage Causes Early and Long-Lasting Microarterial Constriction and Microthrombosis: An in-vivo Microscopy Study. Br. J. Pharmacol. 2011, 32, 447–455. [Google Scholar] [CrossRef]
- Liu, H.; Dienel, A.; Schöller, K.; Schwarzmaier, S.M.; Nehrkorn, K.; Plesnila, N.; Terpolilli, N.A. Microvasospasms After Experimental Subarachnoid Hemorrhage Do Not Depend on Endothelin A Receptors. Stroke 2018, 49, 693–699. [Google Scholar] [CrossRef]
- Tanaka, J.; Seki, Y.; Ishikura, H.; Tsubota, M.; Sekiguchi, F.; Yamaguchi, K.; Murai, A.; Umemura, T.; Kawabata, A. Recombinant human soluble thrombomodulin prevents peripheral HMGB1-dependent hyperalgesia in rats. Br. J. Pharmacol. 2013, 170, 1233–1241. [Google Scholar] [CrossRef]
- Chou, S.H.-Y. Inflammation, Cerebral Vasospasm, and Brain Injury in Subarachnoid Hemorrhage—A Shifting Paradigm and a New Beginning*. Crit. Care Med. 2018, 46, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Inoue, T.; Ohtsuki, S.; Terasaki, T. Quantitative Atlas of Blood–Brain Barrier Transporters, Receptors, and Tight Junction Proteins in Rats and Common Marmoset. J. Pharm. Sci. 2013, 102, 3343–3355. [Google Scholar] [CrossRef] [PubMed]
- Van Zanten, S.E.V.; Hamer, P.C.D.W.; Van Dongen, G.A. Brain Access of Monoclonal Antibodies as Imaged and Quantified by 89Zr-Antibody PET: Perspectives for Treatment of Brain Diseases. J. Nucl. Med. 2019, 60, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.-Y.; Liu, Y.-Z.; Li, J.-M.; Ruan, Y.-M.; Yan, W.-J.; Zhong, S.-Y.; Zhang, T.; Liu, L.-L.; Wu, R.; Wang, B.; et al. Glycyrrhizic acid as an adjunctive treatment for depression through anti-inflammation: A randomized placebo-controlled clinical trial. J. Affect. Disord. 2020, 265, 247–254. [Google Scholar] [CrossRef]


{kind=link}
| Sr. No. | Author/Year | Study Type (Animal Models) | HMGB1 Inhibitor (ip/ICV) | Dose | Key Results |
|---|---|---|---|---|---|
| 1 | An et al. 2018 [34] | Rat Endovascular perforation model | AG 490 | 5 mM in 2 mL DMSO, ICV 30 min before SAH | Reduced apoptosis, edema, improved neurological score |
| 2 | Ieong et al. 2018 [33] | Rat Pre-chiasmatic hemorrhage model | Glycyrrhizin | 15 mg/Kg after SAH, 6 h, 12 h, 18 h, ip | Reduced apoptosis, edema, improved neurological score |
| 3 | Li et al. 2017 [32] | Rat SAH model Double hemorrhage | Glycyrrhizic acid | 10 mg/Kg OD for 3 days, ip | Improved neurologic function, prevented CVS and inflammatory cytokines expression |
| 4 | Zhang et al. 2016 [30] | Pre-chiasmatic hemorrhage model | Resveratrol | 60 mg/Kg in 1% DMSO 2 h and 12 h after SAH, ip | Reduced apoptosis, edema, neurological impairment |
| 5 | Chang et al. 2015 [28] | Rat SAH model Double hemorrhage | 4OGOMV | 100/200/400 µg/Kg/day starting 1 h post SAH for 7 days through mini osmotic pump | Improved CVS, neurological deficits, reduced expression of inflammatory mediators and neuronal apoptosis |
| 6 | Haruma et al. 2016 [38] | Rat SAH model Single hemorrhage | Anti-HMGB1 Antibody | mAb (IgG2a) 1 mg/Kg twice with 24 h interval, iv | Improved CVS, neurological deficits, reduced expression of inflammatory mediators and receptors for vasospastic mediators |
| 7 | Chang et al. 2016 [29] | Rat SAH model Double hemorrhage | Rhinacanthin | 100/200/400 µmol/Kg/day orally in corn oil starting at 1 h after SAH | Reduced apoptosis, improved CVS, neurological deficits, reduced inflammatory mediator expression |
| 8 | Chang et al. 2014 [27] | Rat SAH model Double hemorrhage | Purpurogallin | 100/200/400 µg/Kg/day starting 1 h after SAH through mini osmotic pumps for 5 days | Reduced CVS, inflammatory mediators expression and improved neurological deficits |
| 9 | Wang et al. 2019 [39] | Rat Endovascular perforation model | Anti-HMGB1 Antibody | mAb 1 mg/Kg twice with a 24 h interval after SAH, iv | Reduced CVS, VSMCs phenotype switching & remodelling, brain edema, apoptosis, neurological deficits |
| 10 | Chen et al. 2018 [35] | Rat Endovascular perforation model | GSK 872 | 6 µL of 25 mM GSK 872 in 1% DMSO after 30 min of SAH, ICV | Reduced brain edema, improved neurological scores and reduced neuronal necroptosis |
| 11 | Xiong et al., 2020 [36] | Rat Endovascular perforation model | BMSCs derived exosomes | 1 h after SAH, 200 µg of MSCs-Exo and PBS to final volume of 200 µL, iv | Reduced neurological deficits, brain edema, BBB permeability, mortality, apoptosis and inflammation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, S.; Chaudhry, S.R.; Kahlert, U.D.; Lehecka, M.; Korja, M.; Niemelä, M.; Hänggi, D. Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 2709. https://doi.org/10.3390/ijms21082709
Muhammad S, Chaudhry SR, Kahlert UD, Lehecka M, Korja M, Niemelä M, Hänggi D. Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review. International Journal of Molecular Sciences. 2020; 21(8):2709. https://doi.org/10.3390/ijms21082709
Chicago/Turabian StyleMuhammad, Sajjad, Shafqat Rasul Chaudhry, Ulf Dietrich Kahlert, Martin Lehecka, Miikka Korja, Mika Niemelä, and Daniel Hänggi. 2020. "Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review" International Journal of Molecular Sciences 21, no. 8: 2709. https://doi.org/10.3390/ijms21082709
APA StyleMuhammad, S., Chaudhry, S. R., Kahlert, U. D., Lehecka, M., Korja, M., Niemelä, M., & Hänggi, D. (2020). Targeting High Mobility Group Box 1 in Subarachnoid Hemorrhage: A Systematic Review. International Journal of Molecular Sciences, 21(8), 2709. https://doi.org/10.3390/ijms21082709