Structure and Function of Filamin C in the Muscle Z-Disc


Abstract
1. Introduction
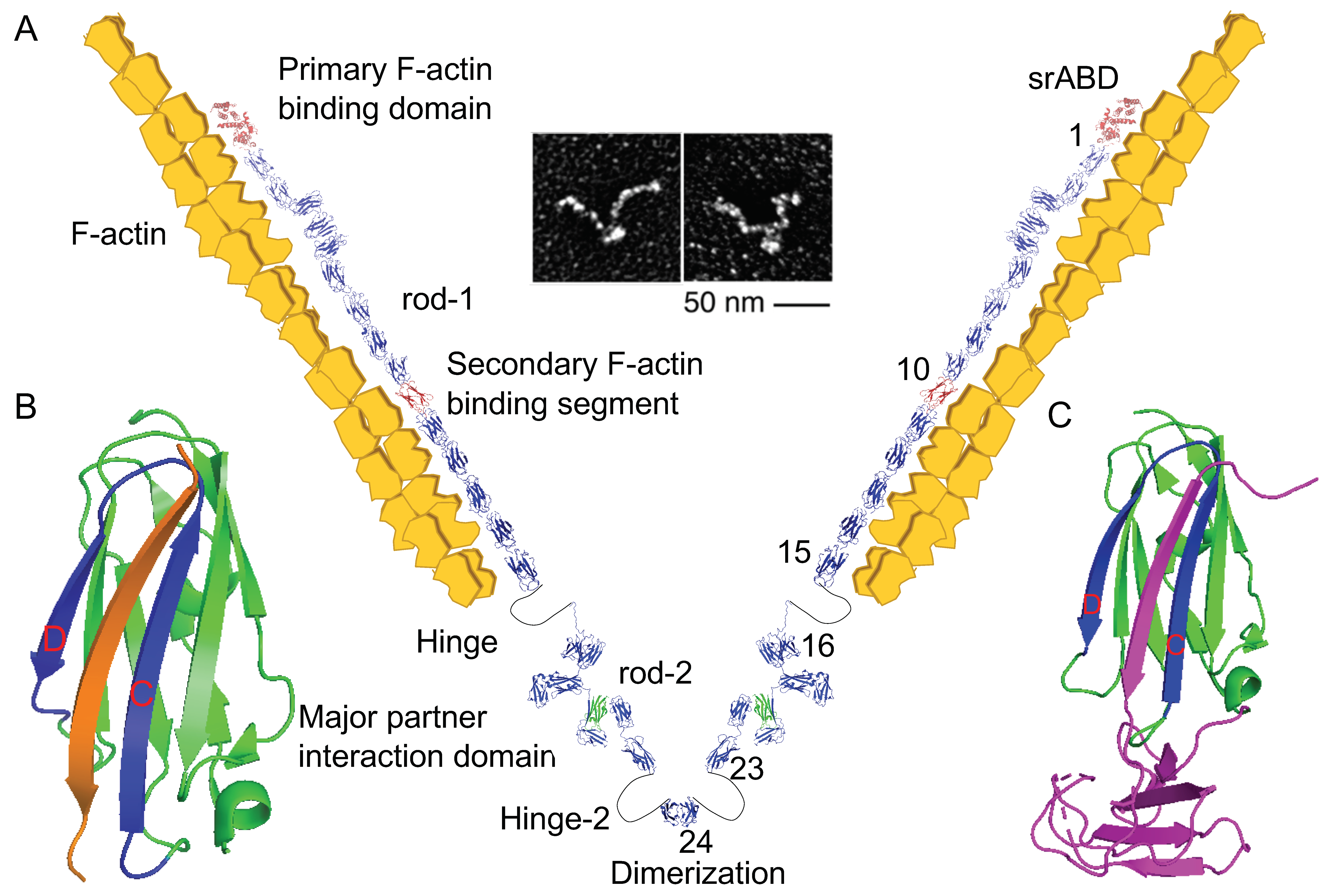
2. Structure of Filamin C (FLNC) and Its Family Proteins
3. FLNC Binding Partners
4. Topology of FLNC in the Z-Disc
5. Association of FLNC Mutations with Myopathies
5.1. FLNC Mutation in Distal and Myofibrillar Skeletal Myopathy
5.2. FLNC Mutation Development in Cardiomyopathy
5.3. Genotype–Phenotype Correlations in FLNC Mutation
6. Post-Translational Modifications
7. Mechanotransduction
8. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FLNC | Filamin C |
| F-actin | Actin filament |
| ABD | Actin-binding domain |
| srABD | Spectrin-related actin-binding domain |
| CH | Calponin homology |
| MFM | Myofibrillar myopathy |
| HCM | Hypertrophic cardiomyopathy |
| DM | Distal myopathy |
| RCM | Restrictive cardiomyopathy |
| DCM | Dilated cardiomyopathy |
| ACM | Arrhythmic cardiomyopathy |
| ARVC | Arrhythmogenic right ventricular cardiomyopathy |
| LGMD | Limb-girdle muscular dystrophy Cardiac arrhythmias |
| CA | Cardiac arrhythmias |
| ABiMVPS | Arrhythmogenic bileaflet mitral valve prolapse syndrome |
| LVNC | Left ventricular non compaction |
References
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adhes. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Maestrini, E.; Patrosso, C.; Mancini, M.; Rivella, S.; Rocchi, M.; Repetto, M.; Villa, A.; Frattini, A.; Zoppe, M.; Vezzoni, P.; et al. Mapping of two genes encoding isoforms of the actin binding protein ABP-280, a dystrophin like protein, to Xq28 and to chromosome 7. Hum. Mol. Genet. 1993, 2, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Xu, W.; Davie, E.W.; Chung, D.W. Molecular cloning of human ABPL, an actin-binding protein homologue. Biochem. Biophys. Res. Commun. 1998, 251, 914–919. [Google Scholar] [CrossRef]
- Thompson, T.G.; Chan, Y.M.; Hack, A.A.; Brosius, M.; Rajala, M.; Lidov, H.G.; McNally, E.M.; Watkins, S.; Kunkel, L.M. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J. Cell Biol. 2000, 148, 115–126. [Google Scholar] [CrossRef]
- Van der Ven, P.F.; Obermann, W.M.; Lemke, B.; Gautel, M.; Weber, K.; Furst, D.O. Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil. Cytoskelet. 2000, 45, 149–162. [Google Scholar] [CrossRef]
- Van der Ven, P.F.; Wiesner, S.; Salmikangas, P.; Auerbach, D.; Himmel, M.; Kempa, S.; Hayess, K.; Pacholsky, D.; Taivainen, A.; Schroder, R.; et al. Indications for a novel muscular dystrophy pathway. gamma-filamin, the muscle-specific filamin isoform, interacts with myotilin. J. Cell Biol. 2000, 151, 235–248. [Google Scholar] [CrossRef]
- Baldassarre, M.; Razinia, Z.; Burande, C.F.; Lamsoul, I.; Lutz, P.G.; Calderwood, D.A. Filamins regulate cell spreading and initiation of cell migration. PLoS ONE 2009, 4, e7830. [Google Scholar] [CrossRef]
- Kesner, B.A.; Milgram, S.L.; Temple, B.R.; Dokholyan, N.V. Isoform divergence of the filamin family of proteins. Mol. Biol. Evol. 2010, 27, 283–295. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, M.H.; Moskowitz, I.P.; Mendonza, A.M.; Vidali, L.; Nakamura, F.; Kwiatkowski, D.J.; Walsh, C.A. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19836–19841. [Google Scholar] [CrossRef]
- Zhou, X.; Tian, F.; Sandzen, J.; Cao, R.; Flaberg, E.; Szekely, L.; Cao, Y.; Ohlsson, C.; Bergo, M.O.; Boren, J.; et al. Filamin B deficiency in mice results in skeletal malformations and impaired microvascular development. Proc. Natl. Acad. Sci. USA 2007, 104, 3919–3924. [Google Scholar] [CrossRef]
- Kyndt, F.; Gueffet, J.P.; Probst, V.; Jaafar, P.; Legendre, A.; Le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 2007, 115, 40–49. [Google Scholar] [CrossRef]
- Furst, D.O.; Goldfarb, L.G.; Kley, R.A.; Vorgerd, M.; Olive, M.; van der Ven, P.F. Filamin C-related myopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, A.A.; Oorschot, V.; Ramm, G.; Bryson-Richardson, R.J. FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Hum. Mol. Genet. 2016, 25, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. FLNC (Filamin-C): A New(er) Player in the Field of Genetic Cardiomyopathies. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2019, in press. [Google Scholar] [CrossRef]
- Ehrlicher, A.J.; Nakamura, F.; Hartwig, J.H.; Weitz, D.A.; Stossel, T.P. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature 2011, 478, 260–263. [Google Scholar] [CrossRef]
- Razinia, Z.; Makela, T.; Ylanne, J.; Calderwood, D.A. Filamins in mechanosensing and signaling. Annu. Rev. Biophys. 2012, 41, 227–246. [Google Scholar] [CrossRef]
- Modarres, H.P.; Mofradt, M.R. Filamin: A structural and functional biomolecule with important roles in cell biology, signaling and mechanics. Mol. Cell. Biomech. 2014, 11, 39–65. [Google Scholar]
- Gariboldi, M.; Maestrini, E.; Canzian, F.; Manenti, G.; De Gregorio, L.; Rivella, S.; Chatterjee, A.; Herman, G.E.; Archidiacono, N.; Antonacci, R.; et al. Comparative mapping of the actin-binding protein 280 genes in human and mouse. Genomics 1994, 21, 428–430. [Google Scholar] [CrossRef]
- Chakarova, C.; Wehnert, M.S.; Uhl, K.; Sakthivel, S.; Vosberg, H.P.; van der Ven, P.F.; Furst, D.O. Genomic structure and fine mapping of the two human filamin gene paralogues FLNB and FLNC and comparative analysis of the filamin gene family. Hum. Genet. 2000, 107, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, A.; Kuikman, I.; Kramer, D.; Geerts, D.; Kreft, M.; Takafuta, T.; Shapiro, S.S.; Sonnenberg, A. Different splice variants of filamin-B affect myogenesis, subcellular distribution, and determine binding to integrin [beta] subunits. J. Cell Biol. 2002, 156, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Korenbaum, E.; Rivero, F. Calponin homology domains at a glance. J Cell Sci 2002, 115, 3543–3545. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Hartwig, J.H.; Stossel, T.P.; Szymanski, P.T. Ca2+ and calmodulin regulate the binding of filamin A to actin filaments. J. Biol. Chem. 2005, 280, 32426–32433. [Google Scholar] [CrossRef]
- Gorlin, J.B.; Yamin, R.; Egan, S.; Stewart, M.; Stossel, T.P.; Kwiatkowski, D.J.; Hartwig, J.H. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): A molecular leaf spring. J. Cell Biol. 1990, 111, 1089–1105. [Google Scholar] [CrossRef]
- Lad, Y.; Kiema, T.; Jiang, P.; Pentikainen, O.T.; Coles, C.H.; Campbell, I.D.; Calderwood, D.A.; Ylanne, J. Structure of three tandem filamin domains reveals auto-inhibition of ligand binding. EMBO J. 2007, 26, 3993–4004. [Google Scholar] [CrossRef]
- Nakamura, F.; Osborn, T.M.; Hartemink, C.A.; Hartwig, J.H.; Stossel, T.P. Structural basis of filamin A functions. J. Cell Biol. 2007, 179, 1011–1025. [Google Scholar] [CrossRef]
- Tossavainen, H.; Koskela, O.; Jiang, P.; Ylanne, J.; Campbell, I.D.; Kilpelainen, I.; Permi, P. Model of a six immunoglobulin-like domain fragment of filamin A (16-21) built using residual dipolar couplings. J. Am. Chem. Soc. 2012, 134, 6660–6672. [Google Scholar] [CrossRef]
- Sethi, R.; Seppala, J.; Tossavainen, H.; Ylilauri, M.; Ruskamo, S.; Pentikainen, O.T.; Pentikainen, U.; Permi, P.; Ylanne, J. A novel structural unit in the N-terminal region of filamins. J. Biol. Chem. 2014, 289, 8588–8598. [Google Scholar] [CrossRef]
- Sethi, R.; Ylanne, J. Small-angle X-ray scattering reveals compact domain-domain interactions in the N-terminal region of filamin C. PLoS ONE 2014, 9, e107457. [Google Scholar] [CrossRef]
- Suphamungmee, W.; Nakamura, F.; Hartwig, J.H.; Lehman, W. Electron microscopy and 3D reconstruction reveals filamin Ig domain binding to F-actin. J. Mol. Biol. 2012, 424, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Sheen, V.L.; Feng, Y.; Graham, D.; Takafuta, T.; Shapiro, S.S.; Walsh, C.A. Filamin A and Filamin B are co-expressed within neurons during periods of neuronal migration and can physically interact. Hum. Mol. Genet. 2002, 11, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.; Van Der Ven, P.F.; Stocklein, W.; Furst, D.O. The limits of promiscuity: Isoform-specific dimerization of filamins. Biochemistry 2003, 42, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Pudas, R.; Heikkinen, O.; Permi, P.; Kilpelainen, I.; Munday, A.D.; Hartwig, J.H.; Stossel, T.P.; Ylanne, J. The structure of the GPIb-filamin A complex. Blood 2006, 107, 1925–1932. [Google Scholar] [CrossRef]
- Wang, J.; Nakamura, F. Identification of Filamin A Mechanobinding Partner II: Fimbacin Is a Novel Actin Cross-Linking and Filamin A Binding Protein. Biochemistry 2019, 58, 4737–4743. [Google Scholar] [CrossRef]
- Wang, L.; Nakamura, F. Identification of Filamin A Mechanobinding Partner I: Smoothelin Specifically Interacts with the Filamin A Mechanosensitive Domain 21. Biochemistry 2019, 58, 4726–4736. [Google Scholar] [CrossRef]
- Nakamura, F.; Heikkinen, O.; Pentikainen, O.T.; Osborn, T.M.; Kasza, K.E.; Weitz, D.A.; Kupiainen, O.; Permi, P.; Kilpelainen, I.; Ylanne, J.; et al. Molecular basis of filamin A-FilGAP interaction and its impairment in congenital disorders associated with filamin A mutations. PLoS ONE 2009, 4, e4928. [Google Scholar] [CrossRef]
- Selcen, D. Myofibrillar myopathies. Neuromuscul. Disord. 2011, 21, 161–171. [Google Scholar] [CrossRef]
- Gontier, Y.; Taivainen, A.; Fontao, L.; Sonnenberg, A.; van der Flier, A.; Carpen, O.; Faulkner, G.; Borradori, L. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J. Cell Sci. 2005, 118, 3739–3749. [Google Scholar] [CrossRef]
- Faulkner, G.; Pallavicini, A.; Comelli, A.; Salamon, M.; Bortoletto, G.; Ievolella, C.; Trevisan, S.; Kojic, S.; Dalla Vecchia, F.; Laveder, P.; et al. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J. Biol. Chem. 2000, 275, 41234–41242. [Google Scholar] [CrossRef]
- Takada, F.; Vander Woude, D.L.; Tong, H.Q.; Thompson, T.G.; Watkins, S.C.; Kunkel, L.M.; Beggs, A.H. Myozenin: An alpha-actinin- and gamma-filamin-binding protein of skeletal muscle Z lines. Proc. Natl. Acad. Sci. USA 2001, 98, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J. Biol. Chem. 2002, 277, 13998–14004. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, A.; van der Ven, P.F.; Vakeel, P.; Albinus, B.; Simonis, D.; Bendas, G.; Schenk, J.A.; Micheel, B.; Kley, R.A.; Furst, D.O. The sarcomeric Z-disc component myopodin is a multiadapter protein that interacts with filamin and alpha-actinin. Eur. J. Cell Biol. 2010, 89, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, J.; Cheng, A.; Deyoung, S.M.; Saltiel, A.R. Identification of CAP as a costameric protein that interacts with filamin C. Mol. Biol. Cell 2007, 18, 4731–4740. [Google Scholar] [CrossRef]
- Van der Ven, P.F.; Ehler, E.; Vakeel, P.; Eulitz, S.; Schenk, J.A.; Milting, H.; Micheel, B.; Furst, D.O. Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp. Cell. Res. 2006, 312, 2154–2167. [Google Scholar] [CrossRef]
- Kley, R.A.; Maerkens, A.; Leber, Y.; Theis, V.; Schreiner, A.; van der Ven, P.F.; Uszkoreit, J.; Stephan, C.; Eulitz, S.; Euler, N.; et al. A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol. Cell. Proteom. 2013, 12, 215–227. [Google Scholar] [CrossRef]
- Eulitz, S.; Sauer, F.; Pelissier, M.C.; Boisguerin, P.; Molt, S.; Schuld, J.; Orfanos, Z.; Kley, R.A.; Volkmer, R.; Wilmanns, M.; et al. Identification of Xin-repeat proteins as novel ligands of the SH3 domains of nebulin and nebulette and analysis of their interaction during myofibril formation and remodeling. Mol. Biol. Cell 2013, 24, 3215–3226. [Google Scholar] [CrossRef]
- Chevessier, F.; Schuld, J.; Orfanos, Z.; Plank, A.C.; Wolf, L.; Maerkens, A.; Unger, A.; Schlotzer-Schrehardt, U.; Kley, R.A.; Von Horsten, S.; et al. Myofibrillar instability exacerbated by acute exercise in filaminopathy. Hum. Mol. Genet. 2015, 24, 7207–7220. [Google Scholar] [CrossRef]
- Yu, J.G.; Furst, D.O.; Thornell, L.E. The mode of myofibril remodelling in human skeletal muscle affected by DOMS induced by eccentric contractions. Histochem. Cell Biol. 2003, 119, 383–393. [Google Scholar] [CrossRef]
- Yu, J.G.; Carlsson, L.; Thornell, L.E. Evidence for myofibril remodeling as opposed to myofibril damage in human muscles with DOMS: An ultrastructural and immunoelectron microscopic study. Histochem. Cell Biol. 2004, 121, 219–227. [Google Scholar] [CrossRef]
- Orfanos, Z.; Godderz, M.P.; Soroka, E.; Godderz, T.; Rumyantseva, A.; van der Ven, P.F.; Hawke, T.J.; Furst, D.O. Breaking sarcomeres by in vitro exercise. Sci. Rep. 2016, 6, 19614. [Google Scholar] [CrossRef]
- Dalkilic, I.; Schienda, J.; Thompson, T.G.; Kunkel, L.M. Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol. Cell. Biol. 2006, 26, 6522–6534. [Google Scholar] [CrossRef] [PubMed]
- Spiro, D. The ultrastructure of striated muscle at various sarcomere lengths. J. Biophys. Biochem. Cytol. 1956, 2 (Suppl. 4), 157–162. [Google Scholar] [CrossRef] [PubMed]
- Knappeis, G.G.; Carlsen, F. The ultrastructure of the Z disc in skeletal muscle. J. Cell Biol. 1962, 13, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Horowits, R. The physiological role of titin in striated muscle. Rev. Physiol. Biochem. Pharmacol. 1999, 138, 57–96. [Google Scholar] [PubMed]
- Huddart, H. The Comparative Structure and Function of Muscle; Pergamon Press: New York, NY, USA, 1975. [Google Scholar]
- Luther, P.K. The vertebrate muscle Z-disc: Sarcomere anchor for structure and signalling. J. Muscle Res. Cell Motil. 2009, 30, 171–185. [Google Scholar] [CrossRef]
- Anastasi, G.; Cutroneo, G.; Trimarchi, F.; Santoro, G.; Bruschetta, D.; Bramanti, P.; Pisani, A.; Favaloro, A. Evaluation of sarcoglycans, vinculin-talin-integrin system and filamin2 in alpha- and gamma-sarcoglycanopathy: An immunohistochemical study. Int. J. Mol. Med. 2004, 14, 989–999. [Google Scholar]
- Leber, Y.; Ruparelia, A.A.; Kirfel, G.; van der Ven, P.F.; Hoffmann, B.; Merkel, R.; Bryson-Richardson, R.J.; Furst, D.O. Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum. Mol. Genet. 2016, 25, 2776–2788. [Google Scholar] [CrossRef]
- Collier, M.P.; Alderson, T.R.; de Villiers, C.P.; Nicholls, D.; Gastall, H.Y.; Allison, T.M.; Degiacomi, M.T.; Jiang, H.; Mlynek, G.; Furst, D.O.; et al. HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci. Adv. 2019, 5, eaav8421. [Google Scholar] [CrossRef]
- Yang, B.; Liu, Y.; Zhao, J.; Hei, K.; Zhuang, H.; Li, Q.; Wei, W.; Chen, R.; Zhang, N.; Li, Y. Ectopic overexpression of filamin C scaffolds MEK1/2 and ERK1/2 to promote the progression of human hepatocellular carcinoma. Cancer Lett. 2017, 388, 167–176. [Google Scholar] [CrossRef]
- Papizan, J.B.; Garry, G.A.; Brezprozvannaya, S.; McAnally, J.R.; Bassel-Duby, R.; Liu, N.; Olson, E.N. Deficiency in Kelch protein Klhl31 causes congenital myopathy in mice. J. Clin. Investig. 2017, 127, 3730–3740. [Google Scholar] [CrossRef] [PubMed]
- Juo, L.Y.; Liao, W.C.; Shih, Y.L.; Yang, B.Y.; Liu, A.B.; Yan, Y.T. HSPB7 interacts with dimerized FLNC and its absence results in progressive myopathy in skeletal muscles. J. Cell Sci. 2016, 129, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Neethling, A.; Mouton, J.; Loos, B.; Corfield, V.; de Villiers, C.; Kinnear, C. Filamin C: A novel component of the KCNE2 interactome during hypoxia. Cardiovasc. J. Afr. 2016, 27, 4–11. [Google Scholar] [CrossRef][Green Version]
- Pawlowski, M.; Saraswathi, S.; Motawea, H.K.; Chotani, M.A.; Kloczkowski, A. In silico modeling of human alpha2C-adrenoreceptor interaction with filamin-2. PLoS ONE 2014, 9, e103099. [Google Scholar] [CrossRef]
- Molt, S.; Buhrdel, J.B.; Yakovlev, S.; Schein, P.; Orfanos, Z.; Kirfel, G.; Winter, L.; Wiche, G.; van der Ven, P.F.; Rottbauer, W.; et al. Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J. Cell Sci. 2014, 127, 3578–3592. [Google Scholar] [CrossRef] [PubMed]
- Spaich, S.; Will, R.D.; Just, S.; Spaich, S.; Kuhn, C.; Frank, D.; Berger, I.M.; Wiemann, S.; Korn, B.; Koegl, M.; et al. F-box and leucine-rich repeat protein 22 is a cardiac-enriched F-box protein that regulates sarcomeric protein turnover and is essential for maintenance of contractile function in vivo. Circ. Res. 2012, 111, 1504–1516. [Google Scholar] [CrossRef]
- Maiweilidan, Y.; Klauza, I.; Kordeli, E. Novel interactions of ankyrins-G at the costameres: The muscle-specific Obscurin/Titin-Binding-related Domain (OTBD) binds plectin and filamin C. Exp. Cell Res. 2011, 317, 724–736. [Google Scholar] [CrossRef]
- Baker, J.; Riley, G.; Romero, M.R.; Haynes, A.R.; Hilton, H.; Simon, M.; Hancock, J.; Tateossian, H.; Ripoll, V.M.; Blanco, G. Identification of a Z-band associated protein complex involving KY, FLNC and IGFN1. Exp. Cell Res. 2010, 316, 1856–1870. [Google Scholar] [CrossRef]
- Nakagawa, K.; Sugahara, M.; Yamasaki, T.; Kajiho, H.; Takahashi, S.; Hirayama, J.; Minami, Y.; Ohta, Y.; Watanabe, T.; Hata, Y.; et al. Filamin associates with stress signalling kinases MKK7 and MKK4 and regulates JNK activation. Biochem. J. 2010, 427, 237–245. [Google Scholar] [CrossRef]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Furst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef]
- Bosch-Comas, A.; Lindsten, K.; Gonzalez-Duarte, R.; Masucci, M.G.; Marfany, G. The ubiquitin-specific protease USP25 interacts with three sarcomeric proteins. Cell. Mol. Life Sci. 2006, 63, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Lahmers, S.; Burkart, C.; Fong, C.; McNabb, M.; Witt, S.; Witt, C.; Labeit, D.; Granzier, H. Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J. Mol. Biol. 2006, 362, 664–681. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Jond-Necand, C.; Marcilhac, A.; Furst, D.; Benyamin, Y. Calpain 1-gamma filamin interaction in muscle cells: A possible in situ regulation by PKC-alpha. Int. J. Biochem. Cell Biol. 2006, 38, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Gainetdinov, R.R.; Laporte, S.A.; Caron, M.G.; Barak, L.S. G protein-coupled receptor kinase regulates dopamine D3 receptor signaling by modulating the stability of a receptor-filamin-beta-arrestin complex. A case of autoreceptor regulation. J. Biol. Chem. 2005, 280, 12774–12780. [Google Scholar] [CrossRef]
- Lypowy, J.; Chen, I.Y.; Abdellatif, M. An alliance between Ras GTPase-activating protein, filamin C, and Ras GTPase-activating protein SH3 domain-binding protein regulates myocyte growth. J. Biol. Chem. 2005, 280, 25717–25728. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.T.; Campbell, D.G.; Peggie, M.; Mora, A.; Cohen, P. Identification of filamin C as a new physiological substrate of PKBalpha using KESTREL. Biochem. J. 2004, 384, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Beatham, J.; Romero, R.; Townsend, S.K.; Hacker, T.; van der Ven, P.F.; Blanco, G. Filamin C interacts with the muscular dystrophy KY protein and is abnormally distributed in mouse KY deficient muscle fibres. Hum. Mol. Genet. 2004, 13, 2863–2874. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, Q.; Chen, F.R.; Han, Q.D.; Zhang, Y.Y. Yeast two-hybrid screening for proteins that interact with alpha1-adrenergic receptors. Acta Pharmacol. Sin. 2004, 25, 1471–1478. [Google Scholar]
- Guyon, J.R.; Kudryashova, E.; Potts, A.; Dalkilic, I.; Brosius, M.A.; Thompson, T.G.; Beckmann, J.S.; Kunkel, L.M.; Spencer, M.J. Calpain 3 cleaves filamin C and regulates its ability to interact with gamma- and delta-sarcoglycans. Muscle Nerve 2003, 28, 472–483. [Google Scholar] [CrossRef]
- Lu, S.; Carroll, S.L.; Herrera, A.H.; Ozanne, B.; Horowits, R. New N-RAP-binding partners alpha-actinin, filamin and Krp1 detected by yeast two-hybrid screening: Implications for myofibril assembly. J. Cell Sci. 2003, 116, 2169–2178. [Google Scholar] [CrossRef]
- Paranavitane, V.; Coadwell, W.J.; Eguinoa, A.; Hawkins, P.T.; Stephens, L. LL5beta is a phosphatidylinositol (3,4,5)-trisphosphate sensor that can bind the cytoskeletal adaptor, gamma-filamin. J. Biol. Chem. 2003, 278, 1328–1335. [Google Scholar] [CrossRef]
- Tigges, U.; Koch, B.; Wissing, J.; Jockusch, B.M.; Ziegler, W.H. The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase C alpha. J. Biol. Chem. 2003, 278, 23561–23569. [Google Scholar] [CrossRef]
- Tu, Y.; Wu, S.; Shi, X.; Chen, K.; Wu, C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 2003, 113, 37–47. [Google Scholar] [CrossRef]
- Ithychanda, S.S.; Das, M.; Ma, Y.Q.; Ding, K.; Wang, X.; Gupta, S.; Wu, C.; Plow, E.F.; Qin, J. Migfilin, a molecular switch in regulation of integrin activation. J. Biol. Chem. 2009, 284, 4713–4722. [Google Scholar] [CrossRef]
- Dyson, J.M.; O′Malley, C.J.; Becanovic, J.; Munday, A.D.; Berndt, M.C.; Coghill, I.D.; Nandurkar, H.H.; Ooms, L.M.; Mitchell, C.A. The SH2-containing inositol polyphosphate 5-phosphatase, SHIP-2, binds filamin and regulates submembraneous actin. J. Cell Biol. 2001, 155, 1065–1079. [Google Scholar] [CrossRef]
- Petrecca, K.; Miller, D.M.; Shrier, A. Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin-binding protein filamin. J. Neurosci. 2000, 20, 8736–8744. [Google Scholar] [CrossRef]
- Brun, F.; Gigli, M.; Graw, S.L.; Judge, D.P.; Merlo, M.; Murray, B.; Calkins, H.; Sinagra, G.; Taylor, M.R.; Mestroni, L.; et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2020, 57, 254–257. [Google Scholar] [CrossRef]
- Hall, C.L.; Gurha, P.; Sabater-Molina, M.; Asimaki, A.; Futema, M.; Lovering, R.C.; Suarez, M.P.; Aguilera, B.; Molina, P.; Zorio, E.; et al. RNA sequencing-based transcriptome profiling of cardiac tissue implicates novel putative disease mechanisms in FLNC-associated arrhythmogenic cardiomyopathy. Int. J. Cardiol. 2020, 302, 124–130. [Google Scholar] [CrossRef]
- Chen, J.; Wu, J.; Han, C.; Li, Y.; Guo, Y.; Tong, X. A mutation in the filamin c gene causes myofibrillar myopathy with lower motor neuron syndrome: A case report. BMC Neurol. 2019, 19, 198. [Google Scholar] [CrossRef]
- Bains, S.; Tester, D.J.; Asirvatham, S.J.; Noseworthy, P.A.; Ackerman, M.J.; Giudicessi, J.R. A Novel Truncating Variant in FLNC-Encoded Filamin C May Serve as a Proarrhythmic Genetic Substrate for Arrhythmogenic Bileaflet Mitral Valve Prolapse Syndrome. Mayo Clin. Proc. 2019, 94, 906–913. [Google Scholar] [CrossRef]
- Roldan-Sevilla, A.; Palomino-Doza, J.; de Juan, J.; Sanchez, V.; Dominguez-Gonzalez, C.; Salguero-Bodes, R.; Arribas-Ynsaurriaga, F. Missense Mutations in the FLNC Gene Causing Familial Restrictive Cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002388. [Google Scholar] [CrossRef]
- Previtali, S.C.; Scarlato, M.; Vezzulli, P.; Ruggieri, A.; Velardo, D.; Benedetti, S.; Torini, G.; Colombo, B.; Maggi, L.; Di Bella, D.; et al. Expanding the central nervous system disease spectrum associated with FLNC mutation. Muscle Nerve 2019, 59, E33–E37. [Google Scholar] [CrossRef]
- Gemelli, C.; Prada, V.; Fiorillo, C.; Fabbri, S.; Maggi, L.; Geroldi, A.; Gibertini, S.; Mandich, P.; Trevisan, L.; Fossa, P.; et al. A novel mutation in the N-terminal acting-binding domain of Filamin C protein causing a distal myofibrillar myopathy. J. Neurol. Sci. 2019, 398, 75–78. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Pu, C.Q.; Ban, R.; Liu, H.X.; Shi, Q.; Lu, X.H. Clinical, Pathological, and Genetic Features of Two Chinese Cases with Filamin C Myopathy. Chin. Med. J. (Engl.) 2018, 131, 2986–2988. [Google Scholar] [CrossRef]
- Ader, F.; De Groote, P.; Reant, P.; Rooryck-Thambo, C.; Dupin-Deguine, D.; Rambaud, C.; Khraiche, D.; Perret, C.; Pruny, J.F.; Mathieu-Dramard, M.; et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin. Genet. 2019, 96, 317–329. [Google Scholar] [CrossRef]
- Cui, H.; Wang, J.; Zhang, C.; Wu, G.; Zhu, C.; Tang, B.; Zou, Y.; Huang, X.; Hui, R.; Song, L.; et al. Mutation profile of FLNC gene and its prognostic relevance in patients with hypertrophic cardiomyopathy. Mol. Genet. Genom. Med. 2018, 6, 1104–1113. [Google Scholar] [CrossRef]
- Sveinbjornsson, G.; Olafsdottir, E.F.; Thorolfsdottir, R.B.; Davidsson, O.B.; Helgadottir, A.; Jonasdottir, A.; Jonasdottir, A.; Bjornsson, E.; Jensson, B.O.; Arnadottir, G.A.; et al. Variants in NKX2-5 and FLNC Cause Dilated Cardiomyopathy and Sudden Cardiac Death. Circ. Genom. Precis. Med. 2018, 11, e002151. [Google Scholar] [CrossRef]
- Schubert, J.; Tariq, M.; Geddes, G.; Kindel, S.; Miller, E.M.; Ware, S.M. Novel pathogenic variants in filamin C identified in pediatric restrictive cardiomyopathy. Hum. Mutat. 2018, 39, 2083–2096. [Google Scholar] [CrossRef]
- Mangum, K.D.; Ferns, S.J. A novel familial truncating mutation in the filamin C gene associated with cardiac arrhythmias. Eur. J. Med. Genet. 2019, 62, 282–285. [Google Scholar] [CrossRef]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated with Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Miao, J.; Su, F.F.; Liu, X.M.; Wei, X.J.; Yuan, Y.; Yu, X.F. A case report: A heterozygous deletion (2791_2805 del) in exon 18 of the filamin C gene causing filamin C-related myofibrillar myopathies in a Chinese family. BMC Neurol. 2018, 18, 79. [Google Scholar] [CrossRef]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef]
- Nozari, A.; Aghaei-Moghadam, E.; Zeinaloo, A.; Mollazadeh, R.; Majnoon, M.T.; Alavi, A.; Ghasemi Firouzabadi, S.; Mohammadzadeh, A.; Banihashemi, S.; Nikzaban, M.; et al. A novel splicing variant in FLNC gene responsible for a highly penetrant familial dilated cardiomyopathy in an extended Iranian family. Gene 2018, 659, 160–167. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, J.; Zhou, Z. Letter by Ma et al Regarding Article, "Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy". Circ. Genom. Precis. Med. 2018, 11, e002117. [Google Scholar] [CrossRef]
- Tucker, N.R.; McLellan, M.A.; Hu, D.; Ye, J.; Parsons, V.A.; Mills, R.W.; Clauss, S.; Dolmatova, E.; Shea, M.A.; Milan, D.J.; et al. Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001780. [Google Scholar] [CrossRef]
- Rossi, D.; Palmio, J.; Evila, A.; Galli, L.; Barone, V.; Caldwell, T.A.; Policke, R.A.; Aldkheil, E.; Berndsen, C.E.; Wright, N.T.; et al. A novel FLNC frameshift and an OBSCN variant in a family with distal muscular dystrophy. PLoS ONE 2017, 12, e0186642. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef]
- Janin, A.; N′Guyen, K.; Habib, G.; Dauphin, C.; Chanavat, V.; Bouvagnet, P.; Eschalier, R.; Streichenberger, N.; Chevalier, P.; Millat, G. Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clin. Genet. 2017, 92, 616–623. [Google Scholar] [CrossRef]
- Van den Bogaart, F.J.; Claeys, K.G.; Kley, R.A.; Kusters, B.; Schrading, S.; Kamsteeg, E.J.; Voermans, N.C. Widening the spectrum of filamin-C myopathy: Predominantly proximal myopathy due to the p.A193T mutation in the actin-binding domain of FLNC. Neuromuscul. Disord. 2017, 27, 73–77. [Google Scholar] [CrossRef]
- Gomez, J.; Lorca, R.; Reguero, J.R.; Moris, C.; Martin, M.; Tranche, S.; Alonso, B.; Iglesias, S.; Alvarez, V.; Diaz-Molina, B.; et al. Screening of the Filamin C Gene in a Large Cohort of Hypertrophic Cardiomyopathy Patients. Circ. Cardiovasc. Genet. 2017, 10, e001584. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated with High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Begay, R.L.; Tharp, C.A.; Martin, A.; Graw, S.L.; Sinagra, G.; Miani, D.; Sweet, M.E.; Slavov, D.B.; Stafford, N.; Zeller, M.J.; et al. FLNC Gene Splice Mutations Cause Dilated Cardiomyopathy. JACC Basic Transl. Sci. 2016, 1, 344–359. [Google Scholar] [CrossRef]
- Avila-Smirnow, D.; Gueneau, L.; Batonnet-Pichon, S.; Delort, F.; Becane, H.M.; Claeys, K.; Beuvin, M.; Goudeau, B.; Jais, J.P.; Nelson, I.; et al. Cardiac arrhythmia and late-onset muscle weakness caused by a myofibrillar myopathy with unusual histopathological features due to a novel missense mutation in FLNC. Rev. Neurol. (Paris) 2016, 172, 594–606. [Google Scholar] [CrossRef]
- Reinstein, E.; Gutierrez-Fernandez, A.; Tzur, S.; Bormans, C.; Marcu, S.; Tayeb-Fligelman, E.; Vinkler, C.; Raas-Rothschild, A.; Irge, D.; Landau, M.; et al. Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. Eur. J. Hum. Genet. 2016, 24, 1792–1796. [Google Scholar] [CrossRef]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Evila, A.; Arumilli, M.; Udd, B.; Hackman, P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul. Disord. 2016, 26, 7–15. [Google Scholar] [CrossRef]
- Dai, Y.; Wei, X.; Zhao, Y.; Ren, H.; Lan, Z.; Yang, Y.; Chen, L.; Cui, L. A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next-generation sequencing. Neuromuscul. Disord. 2015, 25, 617–624. [Google Scholar] [CrossRef]
- Deo, R.C.; Musso, G.; Tasan, M.; Tang, P.; Poon, A.; Yuan, C.; Felix, J.F.; Vasan, R.S.; Beroukhim, R.; De Marco, T.; et al. Prioritizing causal disease genes using unbiased genomic features. Genome Biol. 2014, 15, 534. [Google Scholar] [CrossRef]
- Janssens, J.; Philtjens, S.; Kleinberger, G.; Van Mossevelde, S.; van der Zee, J.; Cacace, R.; Engelborghs, S.; Sieben, A.; Banzhaf-Strathmann, J.; Dillen, L.; et al. Investigating the role of filamin C in Belgian patients with frontotemporal dementia linked to GRN deficiency in FTLD-TDP brains. Acta Neuropathol. Commun. 2015, 3, 68. [Google Scholar] [CrossRef]
- Valdes-Mas, R.; Gutierrez-Fernandez, A.; Gomez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Alvarez, V.; Moris, C.; Leon, D.; et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 5, 5326. [Google Scholar] [CrossRef]
- Tasca, G.; Odgerel, Z.; Monforte, M.; Aurino, S.; Clarke, N.F.; Waddell, L.B.; Udd, B.; Ricci, E.; Goldfarb, L.G. Novel FLNC mutation in a patient with myofibrillar myopathy in combination with late-onset cerebellar ataxia. Muscle Nerve 2012, 46, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Guergueltcheva, V.; Peeters, K.; Baets, J.; Ceuterick-de Groote, C.; Martin, J.J.; Suls, A.; De Vriendt, E.; Mihaylova, V.; Chamova, T.; Almeida-Souza, L.; et al. Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency. Neurology 2011, 77, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Duff, R.M.; Tay, V.; Hackman, P.; Ravenscroft, G.; McLean, C.; Kennedy, P.; Steinbach, A.; Schoffler, W.; van der Ven, P.F.M.; Furst, D.O.; et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am. J. Hum. Genet. 2011, 88, 729–740. [Google Scholar] [CrossRef]
- Luan, X.; Hong, D.; Zhang, W.; Wang, Z.; Yuan, Y. A novel heterozygous deletion-insertion mutation (2695-2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscul. Disord. 2010, 20, 390–396. [Google Scholar] [CrossRef]
- Shatunov, A.; Olive, M.; Odgerel, Z.; Stadelmann-Nessler, C.; Irlbacher, K.; van Landeghem, F.; Bayarsaikhan, M.; Lee, H.S.; Goudeau, B.; Chinnery, P.F.; et al. In-frame deletion in the seventh immunoglobulin-like repeat of filamin C in a family with myofibrillar myopathy. Eur. J. Hum. Genet. 2009, 17, 656–663. [Google Scholar] [CrossRef]
- Vorgerd, M.; van der Ven, P.F.; Bruchertseifer, V.; Lowe, T.; Kley, R.A.; Schroder, R.; Lochmuller, H.; Himmel, M.; Koehler, K.; Furst, D.O.; et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am. J. Hum. Genet. 2005, 77, 297–304. [Google Scholar] [CrossRef]
- Kley, R.A.; Hellenbroich, Y.; van der Ven, P.F.; Furst, D.O.; Huebner, A.; Bruchertseifer, V.; Peters, S.A.; Heyer, C.M.; Kirschner, J.; Schroder, R.; et al. Clinical and morphological phenotype of the filamin myopathy: A study of 31 German patients. Brain 2007, 130, 3250–3264. [Google Scholar] [CrossRef]
- Goldfarb, L.G.; Park, K.Y.; Cervenakova, L.; Gorokhova, S.; Lee, H.S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Hauser, M.A.; Horrigan, S.K.; Salmikangas, P.; Torian, U.M.; Viles, K.D.; Dancel, R.; Tim, R.W.; Taivainen, A.; Bartoloni, L.; Gilchrist, J.M.; et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum. Mol. Genet. 2000, 9, 2141–2147. [Google Scholar] [CrossRef]
- Selcen, D.; Engel, A.G. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann. Neurol. 2005, 57, 269–276. [Google Scholar] [CrossRef]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef]
- Gialluisi, A.; Newbury, D.F.; Wilcutt, E.G.; Olson, R.K.; DeFries, J.C.; Brandler, W.M.; Pennington, B.F.; Smith, S.D.; Scerri, T.S.; Simpson, N.H.; et al. Genome-wide screening for DNA variants associated with reading and language traits. Genes Brain Behav. 2014, 13, 686–701. [Google Scholar] [CrossRef]
- Wilkinson, J.D.; Westphal, J.A.; Bansal, N.; Czachor, J.D.; Razoky, H.; Lipshultz, S.E. Lessons learned from the Pediatric Cardiomyopathy Registry (PCMR) Study Group. Cardiol. Young 2015, 25 (Suppl. 2), 140–153. [Google Scholar] [CrossRef]
- Golbus, J.R.; Puckelwartz, M.J.; Dellefave-Castillo, L.; Fahrenbach, J.P.; Nelakuditi, V.; Pesce, L.L.; Pytel, P.; McNally, E.M. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 751–759. [Google Scholar] [CrossRef]
- Lowe, T.; Kley, R.A.; van der Ven, P.F.; Himmel, M.; Huebner, A.; Vorgerd, M.; Furst, D.O. The pathomechanism of filaminopathy: Altered biochemical properties explain the cellular phenotype of a protein aggregation myopathy. Hum. Mol. Genet. 2007, 16, 1351–1358. [Google Scholar] [CrossRef]
- Kley, R.A.; Serdaroglu-Oflazer, P.; Leber, Y.; Odgerel, Z.; van der Ven, P.F.; Olive, M.; Ferrer, I.; Onipe, A.; Mihaylov, M.; Bilbao, J.M.; et al. Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain 2012, 135, 2642–2660. [Google Scholar] [CrossRef]
- Haataja, T.J.K.; Capoulade, R.; Lecointe, S.; Hellman, M.; Merot, J.; Permi, P.; Pentikainen, U. Critical Structural Defects Explain Filamin A Mutations Causing Mitral Valve Dysplasia. Biophys. J. 2019, 117, 1467–1475. [Google Scholar] [CrossRef]
- Reimann, L.; Wiese, H.; Leber, Y.; Schwable, A.N.; Fricke, A.L.; Rohland, A.; Knapp, B.; Peikert, C.D.; Drepper, F.; van der Ven, P.F.; et al. Myofibrillar Z-discs Are a Protein Phosphorylation Hot Spot with Protein Kinase C (PKCalpha) Modulating Protein Dynamics. Mol. Cell. Proteom. 2017, 16, 346–367. [Google Scholar] [CrossRef]
- Prill, K.; Dawson, J.F. Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases. Int. J. Mol. Sci. 2020, 21, 542. [Google Scholar] [CrossRef]
- Lemke, S.B.; Schnorrer, F. Mechanical forces during muscle development. Mech. Dev. 2017, 144, 92–101. [Google Scholar] [CrossRef]
- Knoll, R.; Buyandelger, B.; Lab, M. The sarcomeric Z-disc and Z-discopathies. J. Biomed. Biotechnol. 2011, 2011, 569628. [Google Scholar] [CrossRef]
- Rognoni, L.; Stigler, J.; Pelz, B.; Ylanne, J.; Rief, M. Dynamic force sensing of filamin revealed in single-molecule experiments. Proc. Natl. Acad. Sci. USA 2012, 109, 19679–19684. [Google Scholar] [CrossRef]
- Kishino, A.; Yanagida, T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature 1988, 334, 74–76. [Google Scholar] [CrossRef]
- Nishizaka, T.; Miyata, H.; Yoshikawa, H.; Ishiwata, S.; Kinosita, K., Jr. Unbinding force of a single motor molecule of muscle measured using optical tweezers. Nature 1995, 377, 251–254. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, X.; Cong, P.; Sheetz, M.P.; Nakamura, F.; Yan, J. Differential mechanical stability of filamin A rod segments. Biophys. J. 2011, 101, 1231–1237. [Google Scholar] [CrossRef]
- Chen, H.; Chandrasekar, S.; Sheetz, M.P.; Stossel, T.P.; Nakamura, F.; Yan, J. Mechanical perturbation of filamin A immunoglobulin repeats 20-21 reveals potential non-equilibrium mechanochemical partner binding function. Sci. Rep. 2013, 3, 1642. [Google Scholar] [CrossRef]
- Xu, T.; Lannon, H.; Wolf, S.; Nakamura, F.; Brujic, J. Domain-domain interactions in filamin A (16-23) impose a hierarchy of unfolding forces. Biophys. J. 2013, 104, 2022–2030. [Google Scholar] [CrossRef]
- Ferrer, J.M.; Lee, H.; Chen, J.; Pelz, B.; Nakamura, F.; Kamm, R.D.; Lang, M.J. Measuring molecular rupture forces between single actin filaments and actin-binding proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9221–9226. [Google Scholar] [CrossRef]
- Molloy, J.E.; Burns, J.E.; Kendrick-Jones, J.; Tregear, R.T.; White, D.C. Movement and force produced by a single myosin head. Nature 1995, 378, 209–212. [Google Scholar] [CrossRef]
- Finer, J.T.; Simmons, R.M.; Spudich, J.A. Single myosin molecule mechanics: Piconewton forces and nanometre steps. Nature 1994, 368, 113–119. [Google Scholar] [CrossRef]
- Haataja, T.J.K.; Bernardi, R.C.; Lecointe, S.; Capoulade, R.; Merot, J.; Pentikainen, U. Non-syndromic Mitral Valve Dysplasia Mutation Changes the Force Resilience and Interaction of Human Filamin A. Structure 2019, 27, 102–112.e4. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Partner | Binding Domain on FLNC | Function | Reference |
|---|---|---|---|
| HSPB1(HSP27) | R18-21 | HspB1, an abundant molecular chaperone and FLNC form a complex. Phosphorylation of HspB1 facilitates extension of FLNC being localized to load-bearing sites. | [60] |
| MEK1/2 ERK1/2 | Co-IP | FLNC enhances the mitogen-activated protein kinase signaling pathway during tumorigenesis. | [61] |
| Klhl31 | Co-IP | Klhl31 targets Flnc for ubiquitination and degradation. | [62] |
| HSPB7 | R24 | Aggregation and mislocalization of FLNC occur in the muscle by loss of HspB7, leading to myopathy. | [63] |
| KCNE2 | Y2H, Co-IP | FLNC and KCNE2, potassium voltage-gated channel, co-localized within the cell, however, a physical interaction was only observed under hypoxic conditions. | [64] |
| α2C-adrenoceptors | 1979 and 2206 (R18-R20) In silico modeling | Phylogenetic and sequence analysis showed that these interactions have evolved in warm-blooded animals. | [65] |
| Aciculin | Co-IP, SPR (R18-21) | Dystrophin-binding protein aciculin interacts FLNC and Xin in Z-line. | [66] |
| Fbxl22 | Co-IP | FLNC is ubiquitinated in Fbxl22-dependent fashion. | [67] |
| Ankyrins-G | R5-6 | Ankyrins-G contains the muscle-specific Obscurin/Titin-Binding-related Domain that binds to FLNC and plectin. | [68] |
| Myopodin (synaptopodin2) | R20-21 | Myopodin also interacts with other Z-line proteins such as alpha-actinin and zyxin. The interaction might play a role in early assembly and stabilization of the Z-disc. | [43] |
| IGFN1 | R19-24 (Y2H) | FLNC interacts IGFN1 and KY at Z-line | [69] |
| MKK4 MKK7 | Co-IP | MKK4 and MKK7 bind all FLNs. FLNA enhances the activation of MKK7 and JNK. | [70] |
| BAG3 | Co-localization | BAG-3 stimulates the release of filamin from a cytoskeleton. Released filamin could subsequently be ubiquitylated by the CHIP/UbcH5 conjugation machinery in the presence of the E1 ubiquitin-activating enzyme. FLNCW2710X blocks BAG3 mediated clearance of protein aggregates. | [14,71] |
| CAP (SORBS1, Ponsin) | R2 | Cbl-associated protein (CAP) is enriched in oxidative muscle fiber. When overexpressed, CAP recruits FLNC to cell-extracellular matrix adhesions and inhibits FLNC-induced cell spreading on fibronectin. | [44] |
| USP25m | Y2H | The ubiquitin-specific protease USP25 interacts with three sarcomeric proteins. | [72] |
| Titin | R20-R24 (Y2H) | Titin Z2-Zis1 domain interacts FLNA/C, alpha-actinin, and nabulin. | [73] |
| Calpain 1 | R23-R24 | Calpain 1 cleaves FLNC hinge-2. Phosphorylation of FLNC by PKC alpha protects the proteolysis of FLNC by calpain 1. | [74] |
| Xin (XIRP1, 2) | R20 (Y2H) | Xin isoforms associate differentially with FLNC. XinB and FLNC compete for binding to aciculin and no ternary complex is formed. | [45,66] |
| β-arrestin2 | R22 (Y2H) | The interaction might regulate dopamine D3 receptor signaling. | [75] |
| RasGAP | R15-R17 | Disrupting the RasGAP-filamin pathway results in reduced myocyte growth. | [76] |
| Integrin beta1A | R20 (Y2H) | [39] | |
| PKBalpha | substrate | PKBalpha phosphoarylate FLNC Ser2213, which lies in an insert not present in the FLNA and FLNB isoforms. Insulin also induced the phosphorylation of FLNC at Ser2213 in cardiac muscle in vivo | [77] |
| KY protein | R20-R22 (Y2H) | KY protein cleaves FLNC. Mutation of KY protein disrupts normal distribution of FLNC. | [78] |
| alpha1-adrenergic receptor | Y2H | Biological significance of the interaction is not known. | [79] |
| Calpain 3 | substrate | FLNC after C3 cleavage, abolishes this interaction with the sarcoglycans. | [80] |
| N-RAP | R20-24 (Y2H) | During myofibril assembly in cultured chick cardiomyocytes, N-RAP, and filamin appear to co-localize with alpha-actinin in the earliest myofibril precursors found near the cell periphery, as well as in the nascent myofibrils that form as these structures fuse laterally. | [81] |
| FLNB | R24 | Heterodimer formation through R24 is possible between FLNC and B but not between FLNA and the other two filamins. | [33] |
| LL5beta | Co-IP | LL5beta binds PI(3,4,5)P3 | [82] |
| PKCalpha | R23-24 (Y2H) | Phosphorylates filamins | [83] |
| Migfilin | R21 | Migfilin interacts with Mig-2 and filamin at cell-matrix adhesion site and regulate cell shape change. | [84,85] |
| SHIP-2 (INPPL1) | R22-23 (Y2H and Co-IP) | Filamin-dependent SHIP-2 localization critically regulates phosphatidylinositol 3 kinase signaling to the actin cytoskeleton. | [86] |
| Myozenin-1, 2, 3 (FATZ, Calsarcins) | R19-24 (Y2H) | Myozenin interacts with FLNC and alpha-actinin in skeletal muscle Z line. | [39,40,41,42] |
| KCND2 | R20-24 (Y2H) | Filamin is required for Kv4.2 localize at filopodial roots. | [87] |
| Myotilin | R20 (Y2H) | Insertion of 82 amino acid residue in R20 defines specific localization of FLNC at Z-line and this domain interacts with myotilin. | [7] |
| γ-, δ-Sarcoglycans | R20-R24 (Y2H) | The identification of FLNC isoform in muscle. | [5,39] |
| Actin | Predicted from sequence similarity and localization in cells. | High homology to actin-binding domains of FLNA and B. | [5] |
| Mutation/Variant | Phenotype | Reference |
|---|---|---|
| c.6565 G>T, p.Glu2189Ter (R20) c.8107delG, p.Asp2703ThrfsTer69 (R24) | Arrhythmogenic right ventricular cardiomyopathy | [88] |
| 7 novel and 2 rare variants | Arrhythmogenic cardiomyopathy | [16,89] |
| heterozygous missense mutation (c.7123G > A, p.V2375I) in R21 | Myofibrillar myopathies with lower motor neuron syndrome | [90] |
| c.201G>A, p.Trp34Ter (srABD) | Arrhythmogenic bileaflet mitral valve prolapse syndrome | [91] |
| c.6902C.T, p.Pro2301Leu (R20) | Familial Restrictive Cardiomyopathy | [92] |
| c.577G > A, p.Ala193Thr (srABD) | Distal and proximal myofibrillar myopathy, cerebellar and CNS sensory ataxia, and pyramidal signs as a consequence of cerebellar and spinal cord abnormalities | [93] |
| c.A664G:p.Met222Val (srABD) | Distal myofibrillar myopathy | [94] |
| p.Asp1691Asn (R15) and p.Asp648Tyr (R4) | myopathy | [95] |
| 28 variants, See the reference | Hypertrophic cardiomyopathies, restrictive cardiomyopathies, dilated cardiomyopathy, left ventricle cardiomyopathy | [96] |
| 43 variants See the reference | Hypertrophic cardiomyopathy | [97] |
| p.Phe1626SerfsTer40 (R14) | Dilated cardiomyopathy with sudden cardiac death | [98] |
| p.Pro2298Leu (R20) p.Tyr2563Cys (R23) | Restrictive cardiomyopathy | [99] |
| c.7536_7548del, p.Pro2513GlufsTer12 (R23) | Cardiac arrhythmias | [100] |
| 6 variants See the reference | Arrhythmogenic dilated cardiomyopathy | [101] |
| c.2791_2805del, p.931_935del (R7) | Myofibrillar myopathies | [102] |
| c.3557C>T, p.Ala1186Val (R10) c.[3547G>C; 3548C>T], p.Ala1183Leu (R10) | Restrictive cardiomyopathy | [103] |
| c.2389+1G>A (exon 15 skipping, stop in R6) | Familial dilated cardiomyopathy | [104] |
| c.6889 G>A, Val2297Met (R20) | Familial Restrictive Cardiomyopathy | [105,106] |
| c.5161delG, p.Gly1722ValfsTer61 (R15) | Distal muscular dystrophy | [107] |
| p.Gly2345Glu (R21) | Congenital heart disease | [108] |
| 10 variants | Dilated cardiomyopathy | [109] |
| c.577G>A, p.Ala193Thr (srABD) | Distal myopathy | [110] |
| 38 variants | Hypertrophic Cardiomyopathy | [111] |
| 23 truncating mutations | Dilated and Arrhythmogenic Cardiomyopathy | [112] |
| c.7251+1 G>A c.5669-1delG | Dilated cardiomyopathy | [113] |
| c.3646T>A, p.Tyr1216Asn (R10) | Myofibrillar myopathy | [114] |
| c.318C>G, p.Phe106Leu (srABD) c.2971C>T, p.Arg991Ter (R8) | Dilated cardiomyopathy | [115] |
| c.4871C>T, p.S1624L (R14) c.6478A>T, p.I2160F (R20) | Familial Restrictive Cardiomyopathy | [116] |
| c.2786-2800del, p.V930-A934del (R7) | Limb-girdle muscular dystrophy | [117] |
| c.969 + 3 A > G | Muscular dystrophy, Congenital myopathy | [118] |
| c.3791 - 1 G>C | Dilated cardiomyopathy | [119] |
| p.V831I (R6) Additional 20 variants | Pick’s disease Frontotemporal dementia | [120] |
| c.4824G>A, p.A1539T (R14) 7 additional mutations | Familial hypertrophic cardiomyopathy | [121] |
| c.7256C>T, p.Thr2419Met (R22) | Myofibrillar myopathy with late-onset cerebellar ataxia | [122] |
| c.5160delC, p.Phe1720LeufsTer63 (R15) | Distal myopathy with upper limb predominance | [123] |
| c.577G>A, p.Ala193Thr (srABD) c.752T>C, p.Met251Thr (srABD) | Distal myopathy | [124] |
| c. 2695-2712 del/GTTTGT ins, p. Lys899-Val904 del, Val899-Cys900 ins (R7) | Myofibrillar myopathy | [125] |
| c.2997–3008del, p.Val930_Thr933del (R7) | Myofibrillar myopathy | [126] |
| c.8130G >A, p.Trp2710Ter (R24) | Myofibrillar myopathy | [14,127,128] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Z.; Nakamura, F. Structure and Function of Filamin C in the Muscle Z-Disc. Int. J. Mol. Sci. 2020, 21, 2696. https://doi.org/10.3390/ijms21082696
Mao Z, Nakamura F. Structure and Function of Filamin C in the Muscle Z-Disc. International Journal of Molecular Sciences. 2020; 21(8):2696. https://doi.org/10.3390/ijms21082696
Chicago/Turabian StyleMao, Zhenfeng, and Fumihiko Nakamura. 2020. "Structure and Function of Filamin C in the Muscle Z-Disc" International Journal of Molecular Sciences 21, no. 8: 2696. https://doi.org/10.3390/ijms21082696
APA StyleMao, Z., & Nakamura, F. (2020). Structure and Function of Filamin C in the Muscle Z-Disc. International Journal of Molecular Sciences, 21(8), 2696. https://doi.org/10.3390/ijms21082696