Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD

 ,
,  and
and 
Abstract
1. Introduction
2. Human Mycobiome
3. Human Virome
4. Human Archaeome
5. Eukaryotic Parasites
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IBD | inflammatory bowel disease |
| CD | Crohn’s disease |
| UC | ulcerative colitis |
| IBS | irritable bowel syndrome |
| SCFA | short-chain fatty acid |
| NGS | next-generation sequencing |
| FMT | fecal microbiota transplantation |
| rCDI | recurrent Clostridium difficile infection |
References
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharm. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Moissl-Eichinger, C.; Probst, A.J.; Birarda, G.; Auerbach, A.; Koskinen, K.; Wolf, P.; Holman, H.Y. Human age and skin physiology shape diversity and abundance of Archaea on skin. Sci. Rep. 2017, 7, 4039. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Sokol, H. The gut mycobiota: Insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastro. Hepat. 2019, 16, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, C.R.; Van der Giezen, M. Associations between Gut Microbiota and Common Luminal Intestinal Parasites. Trends Parasitol. 2018, 34, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef]
- Matijasic, M.; Mestrovic, T.; Peric, M.; Cipcic Paljetak, H.; Panek, M.; Vranesic, B.D.; Ljubas, K.D.; Krznaric, Z.; Verbanac, D. Modulating Composition and Metabolic Activity of the Gut Microbiota in IBD Patients. Int. J. Mol. Sci. 2016, 17, 578. [Google Scholar] [CrossRef]
- Peay, K.G.; Kennedy, P.G.; Talbot, J.M. Dimensions of biodiversity in the Earth mycobiome. Nat. Rev. Microbiol. 2016, 14, 434–447. [Google Scholar] [CrossRef]
- Seed, P.C. The human mycobiome. Cold Spring Harb. Perspect. Med. 2014, 5, a019810. [Google Scholar] [CrossRef]
- Moyes, D.L. The Mycobiome: Influencing IBD Severity. Cell Host Microbe. 2012, 11, 551–552. [Google Scholar] [CrossRef]
- Witherden, E.A.; Shoaie, S.; Hall, R.A.; Moyes, D.L. The Human Mucosal Mycobiome and Fungal Community Interactions. J. Fungi. (Basel) 2017, 3, 56. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Vesty, A.; Biswas, K.; Taylor, M.W.; Gear, K.; Douglas, R.G. Evaluating the Impact of DNA Extraction Method on the Representation of Human Oral Bacterial and Fungal Communities. PLoS ONE 2017, 12, e0169877. [Google Scholar] [CrossRef]
- Diaz, P.I.; Hong, B.Y.; Dupuy, A.K.; Strausbaugh, L.D. Mining the oral mycobiome: Methods, components, and meaning. Virulence 2017, 8, 313–323. [Google Scholar] [CrossRef]
- Motooka, D.; Fujimoto, K.; Tanaka, R.; Yaguchi, T.; Gotoh, K.; Maeda, Y.; Furuta, Y.; Kurakawa, T.; Goto, N.; Yasunaga, T.; et al. Fungal ITS1 Deep-Sequencing Strategies to Reconstruct the Composition of a 26-Species Community and Evaluation of the Gut Mycobiota of Healthy Japanese Individuals. Front. Microbiol. 2017, 8, 238. [Google Scholar] [CrossRef]
- Tang, J.; Iliev, I.D.; Brown, J.; Underhill, D.M.; Funari, V.A. Mycobiome: Approaches to analysis of intestinal fungi. J. Immunol. Methods 2015, 421, 112–121. [Google Scholar] [CrossRef]
- Gdanetz, K.; Benucci, G.M.N.; Vande Pol, N.; Bonito, G. CONSTAX: A tool for improved taxonomic resolution of environmental fungal ITS sequences. BMC Bioinform. 2017, 18, 538. [Google Scholar] [CrossRef]
- Palmer, J.M.; Jusino, M.A.; Banik, M.T.; Lindner, D.L. Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ 2018, 6, e4925. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241. [Google Scholar] [CrossRef]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Laiola, M.; Blaiotta, G.; Ercolini, D. Different Amplicon Targets for Sequencing-Based Studies of Fungal Diversity. Appl. Environ. Microbiol. 2017, 83, e00905–e00917. [Google Scholar] [CrossRef] [PubMed]
- Stielow, J.B.; Levesque, C.A.; Seifert, K.A.; Meyer, W.; Iriny, L.; Smits, D.; Renfurm, R.; Verkley, G.J.M.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 2015, 242–263. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Springer, B.; Pires, V.P.; Keller, P.M. Molecular detection of fungal pathogens in clinical specimens by 18S rDNA high-throughput screening in comparison to ITS PCR and culture. Sci. Rep. 2018, 8, 6964. [Google Scholar] [CrossRef] [PubMed]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef]
- Suhr, M.J.; Hallen-Adams, H.E. The human gut mycobiome: Pitfalls and potentials - a mycologist’s perspective. Mycologia 2015, 107, 1057–1073. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Kachman, S.D.; Kim, J.; Legge, R.M.; Martinez, I. Fungi inhabiting the healthy human gastrointestinal tract: A diverse and dynamic community. Fungal. Ecol. 2015, 15, 9–17. [Google Scholar] [CrossRef]
- Dollive, S.; Chen, Y.Y.; Grunberg, S.; Bittinger, K.; Hoffmann, C.; Vandivier, L.; Cuff, C.; Lewis, J.D.; Wu, G.D.; Bushman, F.D. Fungi of the murine gut: Episodic variation and proliferation during antibiotic treatment. PLoS ONE 2013, 8, e71806. [Google Scholar] [CrossRef]
- Ward, T.L.; Knights, D.; Gale, C.A. Infant fungal communities: Current knowledge and research opportunities. BMC Med. 2017, 15, 30. [Google Scholar] [CrossRef]
- Auchtung, T.A.; Fofanova, T.Y.; Stewart, C.J.; Nash, A.K.; Wong, M.C.; Gesell, J.R.; Auchtung, J.M.; Ajami, N.J.; Petrosino, J.F. Investigating Colonization of the Healthy Adult Gastrointestinal Tract by Fungi. mSphere 2018, 3, e00092-e18. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V. Systematic review and meta-analysis of Saccharomyces boulardii in adult patients. World. J. Gastroenterol 2010, 16, 2202–2222. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V.; Surawicz, C.M.; Greenberg, R.N.; Fekety, R.; Elmer, G.W.; Moyer, K.A.; Melcher, S.A.; Bowen, K.E.; Cox, J.L.; Noorani, Z.; et al. A Randomized Placebo-Controlled Trial of Saccharomyces boulardii in Combination With Standard Antibiotics for Clostridium difficile Disease. JAMA 1994, 271, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Madoff, S.E.; Urquiaga, M.; Alonso, C.D.; Kelly, C.P. Prevention of recurrent Clostridioides difficile infection: A systematic review of randomized controlled trials. Anaerobe 2019, 102098. [Google Scholar] [CrossRef]
- Castagliuolo, I.; Riegler, M.F.; Valenick, L.; LaMont, J.T.; Pothoulakis, C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect. Immun. 1999, 67, 302–307. [Google Scholar] [CrossRef]
- Zbinden, R. Inhibition of Saccharomyces boulardii (nom. inval.) on cell invasion of Salmonella typhimurium and Yersinia enterocolitica. Microb. Ecol. Health Dis. 1999, 11, 158–162. [Google Scholar]
- Thomas, S.; Metzke, D.; Schmitz, J.; Dorffel, Y.; Baumgart, D.C. Anti-inflammatory effects of Saccharomyces boulardii mediated by myeloid dendritic cells from patients with Crohn’s disease and ulcerative colitis. Am. J. Physiol-Gastr. L. 2011, 301, G1083–G1092. [Google Scholar]
- Guslandi, M.; Giollo, P.; Testoni, P.A. A pilot trial of Saccharomyces boulardii in ulcerative colitis. Eur. J. Gastroenterol. Hepatol. 2003, 15, 697–698. [Google Scholar] [CrossRef]
- Guslandi, M.; Mezzi, G.; Sorghi, M.; Testoni, P.A. Saccharomyces boulardii in maintenance treatment of Crohn’s disease. Dig. Dis. Sci. 2000, 45, 1462–1464. [Google Scholar] [CrossRef]
- Qamar, A.; Aboudola, S.; Warny, M.; Michetti, P.; Pothoulakis, C.; LaMont, J.T.; Kelly, C.P. Saccharomyces boulardii stimulates intestinal immunoglobulin A immune response to Clostridium difficile toxin A in mice. Infect. Immun. 2001, 69, 2762–2765. [Google Scholar] [CrossRef]
- Takata, K.; Tomita, T.; Okuno, T.; Kinoshita, M.; Koda, T.; Honorat, J.A.; Takei, M.; Hagihara, K.; Sugimoto, T.; Mochizuki, H.; et al. Dietary Yeasts Reduce Inflammation in Central Nerve System via Microflora. Ann. Clin. Transl. Neurol. 2015, 2, 56–66. [Google Scholar] [CrossRef]
- Mar Rodriguez, M.; Perez, D.; Javier Chaves, F.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jove, M.; Pamplona, R.; Ricart, W.; et al. Obesity changes the human gut mycobiome. Sci. Rep. 2015, 5, 14600. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Botschuijver, S.; Roeselers, G.; Levin, E.; Jonkers, D.M.; Welting, O.; Heinsbroek, S.E.M.; De Weerd, H.H.; Boekhout, T.; Fornai, M.; Masclee, A.A.; et al. Intestinal Fungal Dysbiosis Is Associated With Visceral Hypersensitivity in Patients With Irritable Bowel Syndrome and Rats. Gastroenterology 2017, 153, 1026–1039. [Google Scholar] [CrossRef]
- Costabile, A.; Santarelli, S.; Claus, S.P.; Sanderson, J.; Hudspith, B.N.; Brostoff, J.; Ward, J.L.; Lovegrove, A.; Shewry, P.R.; Jones, H.E.; et al. Effect of Breadmaking Process on In Vitro Gut Microbiota Parameters in Irritable Bowel Syndrome. PLoS ONE 2014, 9, e111225. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.; Yakoob, J.; Jafri, W.; Ahmad, Z.; Azam, Z.; Usman, M.W.; Shamim, S.; Islam, M. Cytokine and clinical response to Saccharomyces boulardii therapy in diarrhea-dominant irritable bowel syndrome: A randomized trial. Eur. J. Gastroen Hepat 2014, 26. [Google Scholar] [CrossRef]
- Main, J.; McKenzie, H.; Yeaman, G.R.; Kerr, M.A.; Robson, D.; Pennington, C.R.; Parratt, D. Antibody to Saccharomyces cerevisiae (bakers’ yeast) in Crohn’s disease. BMJ 1988, 297, 1105–1106. [Google Scholar] [CrossRef]
- Quinton, J.F.; Sendid, B.; Reumaux, D.; Duthilleul, P.; Cortot, A.; Grandbastien, B.; Charrier, G.; Targan, S.R.; Colombel, J.F.; Poulain, D. Anti-Saccharomyces cerevisiae mannan antibodies combined with antineutrophil cytoplasmic autoantibodies in inflammatory bowel disease: Prevalence and diagnostic role. Gut 1998, 42, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Israeli, E.; Grotto, I.; Gilburd, B.; Balicer, R.D.; Goldin, E.; Wiik, A.; Shoenfeld, Y. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut 2005, 54, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Standaert-Vitse, A.; Jouault, T.; Vandewalle, P.; Mille, C.; Seddik, M.; Sendid, B.; Mallet, J.-M.; Colombel, J.-F.; Poulain, D. Candida albicans Is an Immunogen for Anti-Saccharomyces cerevisiae Antibody Markers of Crohns Disease. Gastroenterology 2006, 130, 1764–1775. [Google Scholar] [CrossRef]
- Chehoud, C.; Albenberg, L.G.; Judge, C.; Hoffmann, C.; Grunberg, S.; Bittinger, K.; Baldassano, R.N.; Lewis, J.D.; Bushman, F.D.; Wu, G.D. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. MBio 2016, 7, e01250-16. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; Da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Kuhbacher, T.; Musfeldt, M.; Rosenstiel, P.; Hellmig, S.; Rehman, A.; Drews, O.; Weichert, W.; Timmis, K.N.; Schreiber, S. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand. J. Gastroenterol. 2008, 43, 831–841. [Google Scholar] [CrossRef]
- Qiu, X.; Zhang, F.; Yang, X.; Wu, N.; Jiang, W.; Li, X.; Li, X.; Liu, Y. Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Sci. Rep. 2015, 5, 10416. [Google Scholar] [CrossRef]
- Sovran, B.; Planchais, J.; Jegou, S.; Straube, M.; Lamas, B.; Natividad, J.M.; Agus, A.; Dupraz, L.; Glodt, J.; Da Costa, G.; et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 2018, 6, 152. [Google Scholar] [CrossRef]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immun. 2006, 6, 33–43. [Google Scholar] [CrossRef]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef]
- Tang, C.; Kamiya, T.; Liu, Y.; Kadoki, M.; Kakuta, S.; Oshima, K.; Hattori, M.; Takeshita, K.; Kanai, T.; Saijo, S.; et al. Inhibition of Dectin-1 Signaling Ameliorates Colitis by Inducing Lactobacillus-Mediated Regulatory T Cell Expansion in the Intestine. Cell Host Microbe 2015, 18, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Plato, A.; Hardison, S.E.; Brown, G.D. Pattern recognition receptors in antifungal immunity. Semin. Immunopathol. 2015, 37, 97–106. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Sokol, H.; Conway, K.L.; Zhang, M.; Choi, M.; Morin, B.; Cao, Z.; Villablanca, E.J.; Li, C.; Wijmenga, C.; Yun, S.H.; et al. Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology 2013, 145, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Gessner, M.A.; Werner, J.L.; Lilly, L.M.; Nelson, M.P.; Metz, A.E.; Dunaway, C.W.; Chan, Y.R.; Ouyang, W.; Brown, G.D.; Weaver, C.T.; et al. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect. Immun 2012, 80, 410–417. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Garmaeva, S.; Sinha, T.; Kurilshikov, A.; Fu, J.; Wijmenga, C.; Zhernakova, A. Studying the gut virome in the metagenomic era: Challenges and perspectives. BMC Biol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Lin, D.M.; Lin, H.C. A theoretical model of temperate phages as mediators of gut microbiome dysbiosis. F1000Res 2019, 8, F1000. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Maqsood, R.; Rodgers, R.; Rodriguez, C.; Handley, S.A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Lim, E.S.; Holtz, L.R. Discordant transmission of bacteria and viruses from mothers to babies at birth. Microbiome 2019, 7, 156. [Google Scholar] [CrossRef]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Manrique, P.; Bolduc, B.; Walk, S.T.; Van der Oost, J.; De Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe. 2018, 24, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, R.; Perez-Brocal, V.; Moya, A. Beyond cells-The virome in the human holobiont. Microb. Cell 2019, 6, 373–396. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe. 2019, 26, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The "Known Unknown" of the Microbiome. Cell Host Microbe. 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Ungaro, F.; Massimino, L.; D’Alessio, S.; Danese, S. The gut virome in inflammatory bowel disease pathogenesis: From metagenomics to novel therapeutic approaches. United Eur. Gastroenterol. J. 2019, 7, 999–1007. [Google Scholar] [CrossRef]
- Perez-Brocal, V.; Garcia-Lopez, R.; Nos, P.; Beltran, B.; Moret, I.; Moya, A. Metagenomic Analysis of Crohn’s Disease Patients Identifies Changes in the Virome and Microbiome Related to Disease Status and Therapy, and Detects Potential Interactions and Biomarkers. Inflamm. Bowel Dis. 2015, 21, 2515–2532. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children With Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 30–36. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169. [Google Scholar] [CrossRef]
- Seth, R.K.; Maqsood, R.; Mondal, A.; Bose, D.; Kimono, D.; Holland, L.A.; Janulewicz Lloyd, P.; Klimas, N.; Horner, R.D.; Sullivan, K.; et al. Gut DNA Virome Diversity and Its Association with Host Bacteria Regulate Inflammatory Phenotype and Neuronal Immunotoxicity in Experimental Gulf War Illness. Viruses 2019, 11, 968. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe. 2019, 25, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Kleiner, M.; Paez-Espino, D.; Zhu, W.; Bushnell, B.; Hassell, B.; Winter, S.E.; Kyrpides, N.C.; Hooper, L.V. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol. 2018, 3, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Galtier, M.; Sordi, L.D.; Sivignon, A.; De Vallee, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages Targeting Adherent Invasive Escherichia coli Strains as a Promising New Treatment for Crohn’s Disease. J. Crohn’s Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574. [Google Scholar] [CrossRef]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jovel, J.; Halloran, B.; Wine, E.; Patterson, J.; Ford, G.; O’Keefe, S.; Meng, B.; Song, D.; Zhang, Y.; et al. Metagenomic Analysis of Microbiome in Colon Tissue from Subjects with Inflammatory Bowel Diseases Reveals Interplay of Viruses and Bacteria. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Nahar, S.; Hokama, A.; Fujita, J. Clinical significance of cytomegalovirus and other herpes virus infections in ulcerative colitis. Pol Arch. Intern. Med. 2019, 129, 620–626. [Google Scholar]
- Ungaro, F.; Massimino, L.; Furfaro, F.; Rimoldi, V.; Peyrin-Biroulet, L.; D’Alessio, S.; Danese, S. Metagenomic analysis of intestinal mucosa revealed a specific eukaryotic gut virome signature in early-diagnosed inflammatory bowel disease. Gut Microbes. 2019, 10, 149–158. [Google Scholar] [CrossRef]
- Basic, M.; Keubler, L.M.; Buettner, M.; Achard, M.; Breves, G.; Schroder, B.; Smoczek, A.; Jorns, A.; Wedekind, D.; Zschemisch, N.H.; et al. Norovirus Triggered Microbiota-driven Mucosal Inflammation in Interleukin 10-deficient Mice. Inflamm. Bowel Dis. 2014, 20, 431–443. [Google Scholar] [CrossRef]
- Bolsega, S.; Basic, M.; Smoczek, A.; Buettner, M.; Eberl, C.; Ahrens, D.; Odum, K.A.; Stecher, B.; Bleich, A. Composition of the Intestinal Microbiota Determines the Outcome of Virus-Triggered Colitis in Mice. Front. Immunol. 2019, 10, 1708. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Clooney, A.G.; Hill, C. Giant oversights in the human gut virome. Gut 2019. gutjnl-2019. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.S.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. MBio 2018, 9, e02248-e18. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl Acad Sci USA 1990, 87, 4576. [Google Scholar] [CrossRef] [PubMed]
- Garret, R.; Klenk, H.-P. Archaea: Evolution, Physiology, and Molecular Biology; Blackwell Publishing: Oxford, UK, 2007. [Google Scholar]
- Albers, S.V.; Meyer, B.H. The archaeal cell envelope. Nat. Rev. Microbiol. 2011, 9, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Morii, H. Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol Mol. Biol Rev. 2007, 71, 97–120. [Google Scholar] [CrossRef]
- Valentine, D.L. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat. Rev. Microbiol 2007, 5, 316–323. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Grube, M.; Kaberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2016, 67, 995–1002. [Google Scholar] [CrossRef]
- Janssen, P.H.; Kirs, M. Structure of the Archaeal Community of the Rumen. Appl. Env. Microbiol 2008, 74, 3619. [Google Scholar] [CrossRef]
- Raymann, K.; Moeller, A.H.; Goodman, A.L.; Ochman, H. Unexplored Archaeal Diversity in the Great Ape Gut Microbiome. mSphere 2017, 2, e00026-e17. [Google Scholar] [CrossRef]
- Miller, T.L.; Wolin, M.J.; Conway de Macario, E.; Macario, A.J. Isolation of Methanobrevibacter smithii from human feces. Appl. Env. Microbiol. 1982, 43, 227–232. [Google Scholar] [CrossRef]
- Miller, T.L.; Wolin, M.J. Methanosphaera stadtmaniae gen. nov., sp. nov.: A species that forms methane by reducing methanol with hydrogen. Arch. Microbiol. 1985, 141, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Dridi, B.; Fardeau, M.L.; Ollivier, B.; Raoult, D.; Drancourt, M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2012, 62, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Brusa, T.; Rutili, A.; Canzi, E.; Biavati, B. Isolation and characterization of Methanobrevibacter oralis sp. nov. Curr. Microbiol. 1994, 29, 7–12. [Google Scholar] [CrossRef]
- Nkamga, V.D.; Henrissat, B.; Drancourt, M. Archaea: Essential inhabitants of the human digestive microbiota. Hum. Microbiome, J. 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Wampach, L.; Heintz-Buschart, A.; Hogan, A.; Muller, E.E.L.; Narayanasamy, S.; Laczny, C.C.; Hugerth, L.W.; Bindl, L.; Bottu, J.; Andersson, A.F.; et al. Colonization and Succession within the Human Gut Microbiome by Archaea, Bacteria, and Microeukaryotes during the First Year of Life. Front. Microbiol. 2017, 8, 738. [Google Scholar] [CrossRef]
- Koskinen, K.; Pausan, M.R.; Perras, A.K.; Beck, M.; Bang, C.; Mora, M.; Schilhabel, A.; Schmitz, R.; Moissl-Eichinger, C. First Insights into the Diverse Human Archaeome: Specific Detection of Archaea in the Gastrointestinal Tract, Lung, and Nose and on Skin. MBio 2017, 8, e00824-17. [Google Scholar] [CrossRef]
- Samuel, B.S.; Gordon, J.I. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc. Natl Acad Sci USA 2006, 103, 10011. [Google Scholar] [CrossRef]
- Chaudhary, P.P.; Conway, P.L.; Schlundt, J. Methanogens in humans: Potentially beneficial or harmful for health. Appl. Microbiol. Biotechnol. 2018, 102, 3095–3104. [Google Scholar] [CrossRef]
- Dridi, B.; Henry, M.; El Khechine, A.; Raoult, D.; Drancourt, M. High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS ONE 2009, 4, e7063. [Google Scholar] [CrossRef]
- Samuel, B.S.; Hansen, E.E.; Manchester, J.K.; Coutinho, P.M.; Henrissat, B.; Fulton, R.; Latreille, P.; Kim, K.; Wilson, R.K.; Gordon, J.I. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc. Natl. Acad. Sci. USA 2007, 104, 10643–10648. [Google Scholar] [CrossRef] [PubMed]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.W.; Brugere, J.F. Archaea and the human gut: New beginning of an old story. World. J. Gastroenterol. 2014, 20, 16062–16078. [Google Scholar] [CrossRef] [PubMed]
- Khelaifia, S.; Caputo, A.; Andrieu, C.; Cadoret, F.; Armstrong, N.; Michelle, C.; Lagier, J.C.; Djossou, F.; Fournier, P.E.; Raoult, D. Genome sequence and description of Haloferax massiliense sp. nov., a new halophilic archaeon isolated from the human gut. Extremophiles 2018, 22, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Khelaifia, S.; Raoult, D. Haloferax massiliensis sp. nov., the first human-associated halophilic archaea. New Microbes. New Infect. 2016, 12, 96–98. [Google Scholar] [CrossRef]
- Bang, C.; Weidenbach, K.; Gutsmann, T.; Heine, H.; Schmitz, R.A. The Intestinal Archaea Methanosphaera stadtmanae and Methanobrevibacter smithii Activate Human Dendritic Cells. PLoS ONE 2014, 9, e99411. [Google Scholar] [CrossRef]
- Blais Lecours, P.; Duchaine, C.; Taillefer, M.; Tremblay, C.; Veillette, M.; Cormier, Y.; Marsolais, D. Immunogenic properties of archaeal species found in bioaerosols. PLoS ONE 2011, 6, e23326. [Google Scholar] [CrossRef]
- Bang, C.; Vierbuchen, T.; Gutsmann, T.; Heine, H.; Schmitz, R.A. Immunogenic properties of the human gut-associated archaeon Methanomassiliicoccus luminyensis and its susceptibility to antimicrobial peptides. PLoS ONE 2017, 12, e0185919. [Google Scholar] [CrossRef]
- Borrel, G.; McCann, A.; Deane, J.; Neto, M.C.; Lynch, D.B.; Brugere, J.F.; O’Toole, P.W. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome. Isme J. 2017, 11, 2059–2074. [Google Scholar] [CrossRef]
- Liu, T.X.; Niu, H.T.; Zhang, S.Y. Intestinal Microbiota Metabolism and Atherosclerosis. Chin. Med. J. (Engl) 2015, 128, 2805–2811. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Brugere, J.F.; Borrel, G.; Gaci, N.; Tottey, W.; O’Toole, P.W.; Malpuech-Brugere, C. Archaebiotics: Proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes 2014, 5, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Tottey, W.; Feria-Gervasio, D.; Gaci, N.; Laillet, B.; Pujos, E.; Martin, J.F.; Sebedio, J.L.; Sion, B.; Jarrige, J.F.; Alric, M.; et al. Colonic Transit Time Is a Driven Force of the Gut Microbiota Composition and Metabolism: In Vitro Evidence. J. Neurogastroenterol. Motil 2017, 23, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllou, K.; Chang, C.; Pimentel, M. Methanogens, methane and gastrointestinal motility. J. Neurogastroenterol. Motil. 2014, 20, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, D.; Ghosh, S.; Dutta, T.K.; Ahn, Y. Current State of Knowledge in Microbial Degradation of Polycyclic Aromatic Hydrocarbons (PAHs): A Review. Front. Microbiol. 2016, 7, 1369. [Google Scholar] [CrossRef]
- Hubert, S.; Chadwick, A.; Wacher, V.; Coughlin, O.; Kokai-Kun, J.; Bristol, A. Development of a Modified-Release Formulation of Lovastatin Targeted to Intestinal Methanogens Implicated in Irritable Bowel Syndrome With Constipation. J. Pharm. Sci. 2018, 107, 662–671. [Google Scholar] [CrossRef]
- Blais Lecours, P.; Marsolais, D.; Cormier, Y.; Berberi, M.; Hache, C.; Bourdages, R.; Duchaine, C. Increased Prevalence of Methanosphaera stadtmanae in Inflammatory Bowel Diseases. PLoS ONE 2014, 9, e87734. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Marchesi, J.R. Micro-eukaryotic diversity of the human distal gut microbiota: Qualitative assessment using culture-dependent and -independent analysis of faeces. Isme J. 2008, 2, 1183–1193. [Google Scholar] [CrossRef]
- Ghavami, S.B.; Rostami, E.; Sephay, A.A.; Shahrokh, S.; Balaii, H.; Aghdaei, H.A.; Zali, M.R. Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb. Pathog. 2018, 117, 285–289. [Google Scholar] [CrossRef]
- Burman, S.; Hoedt, E.C.; Pottenger, S.; Mohd-Najman, N.S.; O Cuiv, P.; Morrison, M. An (Anti)-Inflammatory Microbiota: Defining the Role in Inflammatory Bowel Disease? Dig. Dis. 2016, 34, 64–71. [Google Scholar] [CrossRef]
- White, J.F. Syntrophic imbalance and the etiology of bacterial endoparasitism diseases. Med. Hypotheses 2017, 107, 14–15. [Google Scholar] [CrossRef]
- Pimentel, M.; Gunsalus, R.P.; Rao, S.S.; Zhang, H. Methanogens in Human Health and Disease. Am. J. Gastroenterol. Suppl. 2012, 1, 28–33. [Google Scholar] [CrossRef]
- Goncalves, P.; Araujo, J.R.; Di Santo, J.P. A Cross-Talk Between Microbiota-Derived Short-Chain Fatty Acids and the Host Mucosal Immune System Regulates Intestinal Homeostasis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef]
- Zaleski, A.; Banaszkiewicz, A.; Walkowiak, J. Butyric acid in irritable bowel syndrome. Prz. Gastroenterol. 2013, 8, 350–353. [Google Scholar] [PubMed]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; De Vos, M.; Boon, N.; Van de Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 11450. [Google Scholar] [CrossRef] [PubMed]
- White, J.F.; Kingsley, K.L.; Verma, S.K.; Kowalski, K.P. Rhizophagy Cycle: An Oxidative Process in Plants for Nutrient Extraction from Symbiotic Microbes. Microorganisms 2018, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Grine, G.; Boualam, M.A.; Drancourt, M. Methanobrevibacter smithii, a methanogen consistently colonising the newborn stomach. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2449–2455. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2015, 1, e00009–e00015. [Google Scholar] [CrossRef]
- Fischer, M.A.; Gullert, S.; Neulinger, S.C.; Streit, W.R.; Schmitz, R.A. Evaluation of 16S rRNA Gene Primer Pairs for Monitoring Microbial Community Structures Showed High Reproducibility within and Low Comparability between Datasets Generated with Multiple Archaeal and Bacterial Primer Pairs. Front. Microbiol. 2016, 7, 1297. [Google Scholar] [CrossRef]
- Mahnert, A.; Blohs, M.; Pausan, M.R.; Moissl-Eichinger, C. The human archaeome: Methodological pitfalls and knowledge gaps. Emerg. Top. Life Sci. 2018, 2, 469–482. [Google Scholar]
- Pausan, M.R.; Csorba, C.; Singer, G.; Till, H.; Sch+Âpf, V.; Santigli, E.; Klug, B.; Hagenauer, C.; Blohs, M.; Moissl-Eichinger, C. Exploring the Archaeome: Detection of Archaeal Signatures in the Human Body. Front. Microbiol. 2019, 10, 2796. [Google Scholar] [CrossRef]
- Eichenberger, R.M.; Ryan, S.; Jones, L.; Buitrago, G.; Polster, R.; Montes de Oca, M.; Zuvelek, J.; Giacomin, P.R.; Dent, L.A.; Engwerda, C.R.; et al. Hookworm Secreted Extracellular Vesicles Interact With Host Cells and Prevent Inducible Colitis in Mice. Front. Immunol. 2018, 9, 850. [Google Scholar] [CrossRef]
- Hamad, I.; Raoult, D.; Bittar, F. Repertory of eukaryotes (eukaryome) in the human gastrointestinal tract: Taxonomy and detection methods. Parasite Immunol. 2016, 38, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Mirjalali, H.; Abbasi, M.R.; Naderi, N.; Hasani, Z.; Mirsamadi, E.S.; Stensvold, C.R.; Balaii, H.; Asadzadeh, A.H.; Zali, M.R. Distribution and phylogenetic analysis of Blastocystis sp. subtypes isolated from IBD patients and healthy individuals in Iran. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Tito, R.Y.; Chaffron, S.; Caenepeel, C.; Lima-Mendez, G.; Wang, J.; Vieira-Silva, S.; Falony, G.; Hildebrand, F.; Darzi, Y.; Rymenans, L.; et al. Population-level analysis of Blastocystis subtype prevalence and variation in the human gut microbiota. Gut 2019, 68, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Furusho, J.K.; Torijano-Carrera, E. Intestinal protozoa infections among patients with ulcerative colitis: Prevalence and impact on clinical disease course. Digestion. 2010, 82, 18–23. [Google Scholar] [CrossRef]
- Tai, W.P.; Hu, P.J.; Wu, J.; Lin, X.C. Six ulcerative colitis patients with refractory symptoms co-infective with Blastocystis hominis in China. Parasitol. Res. 2011, 108, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Yason, J.A.; Liang, Y.R.; Png, C.W.; Zhang, Y.; Tan, K.S.W. Interactions between a pathogenic Blastocystis subtype and gut microbiota: In vitro and in vivo studies. Microbiome 2019, 7, 30. [Google Scholar] [CrossRef]
- Nourrisson, C.; Scanzi, J.; Pereira, B.; NkoudMongo, C.; Wawrzyniak, I.; Cian, A.; Viscogliosi, E.; Livrelli, V.; Delbac, F.; Dapoigny, M.; et al. Blastocystis is associated with decrease of fecal microbiota protective bacteria: Comparative analysis between patients with irritable bowel syndrome and control subjects. PLoS ONE 2014, 9, e111868. [Google Scholar] [CrossRef]
- Petersen, A.M.; Stensvold, C.R.; Mirsepasi, H.; Engberg, J.; Friis-Moller, A.; Porsbo, L.J.; Hammerum, A.M.; Nordgaard-Lassen, I.; Nielsen, H.V.; Krogfelt, K.A. Active ulcerative colitis associated with low prevalence of Blastocystis and Dientamoeba fragilis infection. Scand. J. Gastroenterol. 2013, 48, 638–639. [Google Scholar] [CrossRef]
- Rossen, N.G.; Bart, A.; Verhaar, N.; Van Nood, E.; Kootte, R.; De Groot, P.F.; D’Haens, G.R.; Ponsioen, C.Y.; Van Gool, T. Low prevalence of Blastocystis sp. in active ulcerative colitis patients. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1039–1044. [Google Scholar] [CrossRef]
- Coskun, A.; Malatyali, E.; Ertabaklar, H.; Yasar, M.B.; Karaoglu, A.O.; Ertug, S. Blastocystis in ulcerative colitis patients: Genetic diversity and analysis of laboratory findings. Asian Pac. J. Trop. Med. 2016, 9, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Audebert, C.; Even, G.; Cian, A.; Blastocystis Investigation Group; Loywick, A.; Merlin, S.; Viscogliosi, E.; Chabe, M. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci. Rep. 2016, 6, 25255. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.; Cekin, Y.; Cekin, A.H.; Uyar, S.; Harmandar, F.; Sahinturk, Y. The role of Blastocystis hominis in the activation of ulcerative colitis. Turk. J. Gastroenterol. 2019, 30, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard, L.R.; Engsbro, A.L.; Stensvold, C.R.; Nielsen, H.V.; Bytzer, P. The prevalence of intestinal parasites is not greater among individuals with irritable bowel syndrome: A population-based case-control study. Clin. Gastroenterol. Hepatol. 2015, 13, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard, L.R.; Andersen, L.O.; Johannesen, T.B.; Engsbro, A.L.; Stensvold, C.R.; Nielsen, H.V.; Bytzer, P. Characteristics of the bacterial microbiome in association with common intestinal parasites in irritable bowel syndrome. Clin. Transl. Gastroenterol. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Sipahi, A.M.; Baptista, D.M. Helminths as an alternative therapy for intestinal diseases. World. J. Gastroenterol.. 2017, 23, 6009–6015. [Google Scholar] [CrossRef]
- Lukes, J.; Kuchta, R.; Scholz, T.; Pomajbikova, K. (Self-) infections with parasites: Re-interpretations for the present. Trends Parasitol. 2014, 30, 377–385. [Google Scholar] [CrossRef]
- Terveer, E.M.; Van Gool, T.; Ooijevaar, R.E.; Sanders, I.M.J.G.; Boeije-Koppenol, E.; Keller, J.J.; Bart, A.; Kuijper, E.J.; Netherlands Donor Feces Bank (NDFB) Study Group. Human Transmission of Blastocystis by Fecal Microbiota Transplantation Without Development of Gastrointestinal Symptoms in Recipients. Clin. Infect. Dis. 2019. [Epub ahead of print]. [Google Scholar] [CrossRef]
- Joos, L.; Loosli, J.; Spichtin, H.P.; Krause, M. Amoebic liver abscess in chronic colitis: Revision of “Crohn disease” diagnosis. Schweiz Med. Wochenschr. 1999, 129, 1656–1659. [Google Scholar]
- Verstockt, B.; Vermeire, S.; Van Assche, G.; Ferrante, M. When IBD is not IBD. Scand. J. Gastroenterol. 2018, 53, 1085–1088. [Google Scholar] [CrossRef]
- Vadlamudi, N.; Maclin, J.; Dimmitt, R.A.; Thame, K.A. Cryptosporidial infection in children with inflammatory bowel disease. J. Crohns Colitis 2013, 7, e337–e343. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, A.; Zorzi, F.; Del Chierico, F.; Altomare, A.; Cocca, S.; Avola, A.; De Biasio, F.; Russo, A.; Cella, E.; Reddel, S.; et al. Fecal and Mucosal Microbiota Profiling in Irritable Bowel Syndrome and Inflammatory Bowel Disease. Front. Microbiol. 2019, 10, 1655. [Google Scholar] [CrossRef] [PubMed]
- Morton, E.R.; Lynch, J.; Froment, A.; Lafosse, S.; Heyer, E.; Przeworski, M.; Blekhman, R.; S+ęgurel, L. Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genet. 2015, 11, e1005658. [Google Scholar] [CrossRef] [PubMed]
- Creased sampling reveals novel lineages of Entamoeba: Consequences of genetic diversity and host specificity for taxonomy and molecular detection. Protist 2011, 162, 525–541. [CrossRef] [PubMed]


{kind=link}
{kind=link}
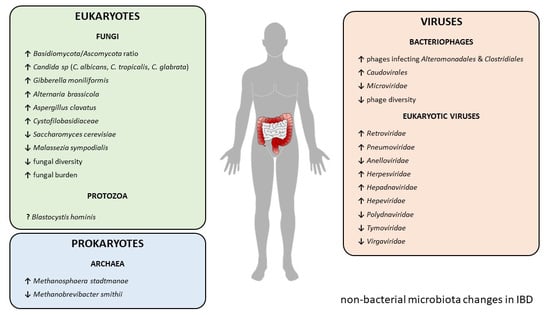
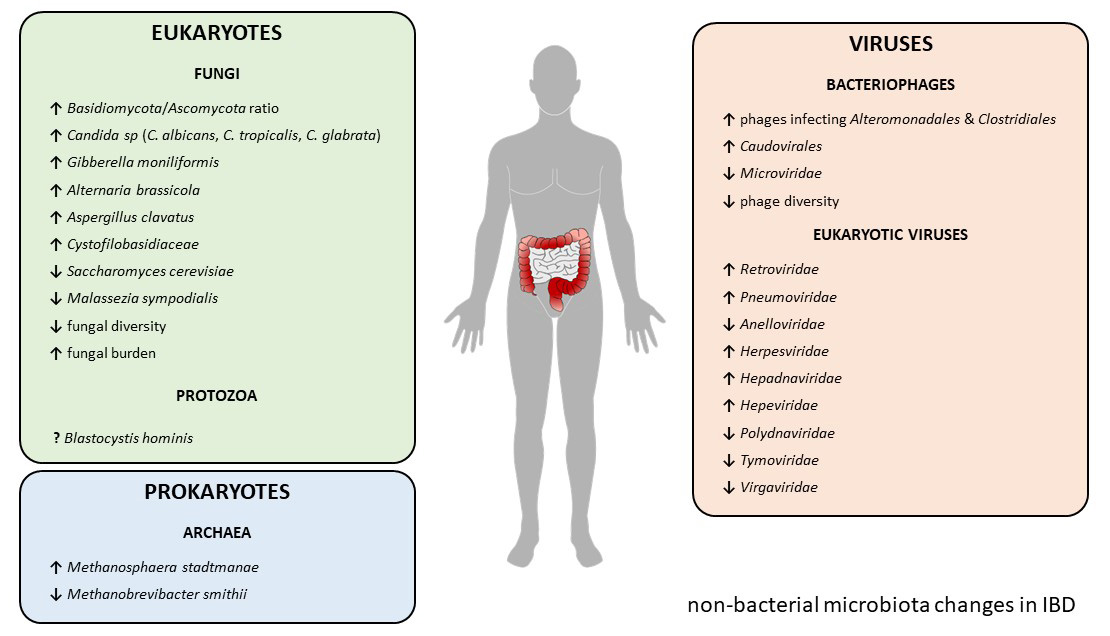
| IBD Type | Change | Reference | ||
|---|---|---|---|---|
| Mycobiome | CD + UC | ↑ Basidiomycota/Ascomycota ratio | [53] | |
| CD + UC | ↑ Candida albicans | [53] | ||
| CD | ↑ Candida tropicalis | [54] | ||
| CD | ↑ Candida glabrata | [55] | ||
| CD | ↑ Gibberella moniliformis | [55] | ||
| CD | ↑ Alternaria brassicola | [55] | ||
| CD | ↑ Aspregillus clavatus | [55] | ||
| CD | ↑ Cystofilobasidiaceae family | [55] | ||
| CD + UC | ↓ Saccharomyces cerevisiae | [53] | ||
| CD + UC | ↓ Malassezia sympodialis | [53] | ||
| UC | ↓ Fungal diversity | [53] | ||
| CD + UC | ↑ Fungal burden | [55,56] | ||
| UC | ↑ Fungal–bacteria interactions | [53] | ||
| CD | ↓ Fungal–bacteria interactions | [53] | ||
| Virome | Phageome | CD | ↑ Phages infecting bacterial orders Alteronomoadales and Clostridiales | [87] |
| CD | ↓ Microviridae family | [89] | ||
| CD + UC | ↑ Caudovirales order | [88,90] | ||
| CD + UC | ↓ Phage diversity | [88,90] | ||
| Eukaryotic virome | CD | ↑ Retroviridae family | [87] | |
| UC | ↑ Pneumoviridae family | [90] | ||
| UC | ↓ Anelloviridae family | [90] | ||
| CD + UC | ↑ Herpesviridae family | [97,98] | ||
| CD + UC | ↑ Hepadnaviridae family | [99] | ||
| CD + UC | ↑ Hepeviridae family | [99] | ||
| UC | ↓ Polydnaviridae family | [99] | ||
| UC | ↓ Tymoviridae family | [99] | ||
| CD | ↓ Virgaviridae family | [99] | ||
| Archaeome | CD + UC | ↓ Methanobrevibacter smithii | [138,140] | |
| CD + UC | ↑ Methanosphaera stadtmanae | [138] | ||
| Eukaryotic parasites | UC | ↑ Blastocystis hominis | [157,158] | |
| UC | ↓ Blastocystis hominis | [161,162,163,164,165] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matijašić, M.; Meštrović, T.; Čipčić Paljetak, H.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. https://doi.org/10.3390/ijms21082668
Matijašić M, Meštrović T, Čipčić Paljetak H, Perić M, Barešić A, Verbanac D. Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. International Journal of Molecular Sciences. 2020; 21(8):2668. https://doi.org/10.3390/ijms21082668
Chicago/Turabian StyleMatijašić, Mario, Tomislav Meštrović, Hana Čipčić Paljetak, Mihaela Perić, Anja Barešić, and Donatella Verbanac. 2020. "Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD" International Journal of Molecular Sciences 21, no. 8: 2668. https://doi.org/10.3390/ijms21082668
APA StyleMatijašić, M., Meštrović, T., Čipčić Paljetak, H., Perić, M., Barešić, A., & Verbanac, D. (2020). Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. International Journal of Molecular Sciences, 21(8), 2668. https://doi.org/10.3390/ijms21082668