Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular



 , ,
, , 


{kind=link}
Abstract
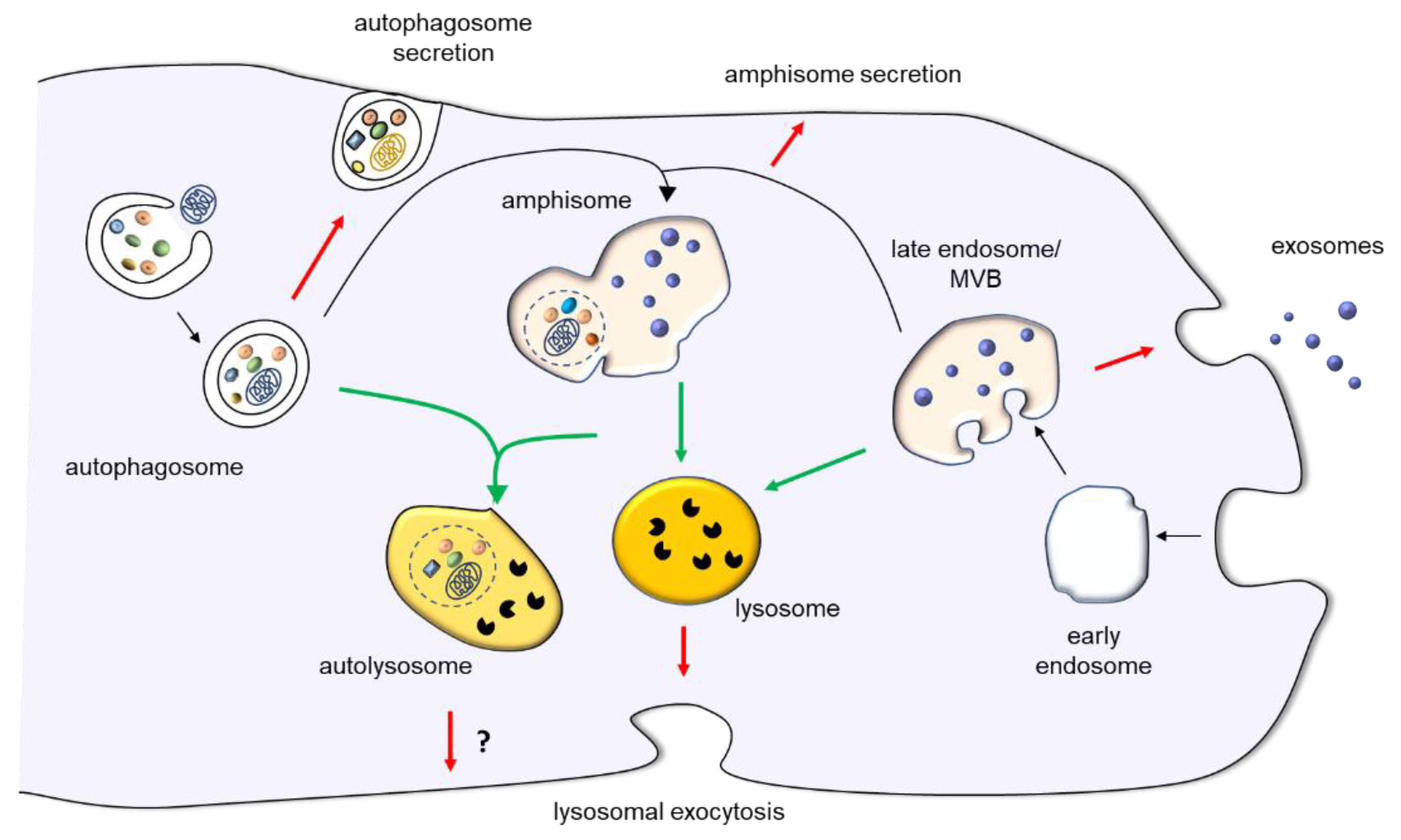
1. Introduction
2. Lysosomal Exocytosis
3. Exosomes
4. Secretory Autophagy
4.1. Autophagy: An Overview
4.2. The Autophagic Processes
4.3. Non Degradative Function of Autophagy: Secretory Autophagy
4.4. The Molecular Machinery underlying Secretory Autophagy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [PubMed]
- Lettau, M.; Schmidt, H.; Kabelitz, D.; Janssen, O. Secretory lysosomes and their cargo in T and NK cells. Immunol. Lett. 2007, 108, 10–19. [Google Scholar] [PubMed]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [PubMed]
- Corrotte, M.; Castro-Gomes, T. Lysosomes and plasma membrane repair. Curr. Top. Membr. 2019, 84, 1–16. [Google Scholar]
- Mostov, K.; Werb, Z. Journey across the osteoclast. Science 1997, 276, 219–220. [Google Scholar]
- Stinchcombe, J.; Bossi, G.; Griffiths, G.M. Linking albinism and immunity: The secrets of secretory lysosomes. Science 2004, 305, 55–59. [Google Scholar]
- Bossi, G.; Griffiths, G.M. CTL secretory lysosomes: Biogenesis and secretion of a harmful organelle. Semin. Immunol. 2005, 17, 87–94. [Google Scholar]
- Logan, M.R.; Odemuyiwa, S.O.; Moqbel, R. Understanding exocytosis in immune and inflammatory cells: The molecular basis of mediator secretion. J. Allergy Clin. Immunol. 2003, 111, 923–932. [Google Scholar]
- Zhang, Z.; Chen, G.; Zhou, W.; Song, A.; Xu, T.; Luo, Q.; Wang, W.; Gu, X.S.; Duan, S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 2007, 9, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Neary, J. P2X7/P2Z receptors: Properties and relevance to CNS functions. Glia 2006, 54, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gu, Y.; Wen, R.; Shen, F.; Tian, H.L.; Yang, G.Y.; Zhang, Z. Lysosome exocytosis is involved in astrocyte ATP release after oxidative stress induced by H2O2. Neurosci. Lett. 2019, 705, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef]
- Andrews, N.W. Membrane resealing: Synaptotagmin VII keeps running the show. Sci. Signal. 2005, 2005, pe19. [Google Scholar] [CrossRef]
- Czibener, C.; Sherer, N.M.; Becker, S.M.; Pypaert, M.; Hui, E.; Chapman, E.R.; Mothes, W.; Andrews, N.W. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol. 2006, 174, 997–1007. [Google Scholar] [CrossRef]
- Arantes, R.M.; Andrews, N.W. A role for synaptotagmin VII-regulated exocytosis of lysosomes in neurite outgrowth from primary sympathetic neurons. J. Neurosci. 2006, 26, 4630–4637. [Google Scholar] [CrossRef]
- Naegeli, K.M.; Hastie, E.; Garde, A.; Wang, Z.; Keeley, D.P.; Gordon, K.L.; Pani, A.M.; Kelley, L.C.; Morrissey, M.A.; Chi, Q.; et al. Cell Invasion In Vivo via Rapid Exocytosis of a Transient Lysosome-Derived Membrane Domain. Dev. Cell 2017, 43, 403–417. [Google Scholar] [CrossRef]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Cyphersmith, A.; Zapata, J.A.; Kim, Y.J.; Payne, C.K. Lysosome transport as a function of lysosome diameter. PLoS ONE 2014, 9, e86847. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.K.; Huynh, C. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef] [PubMed]
- Tucker, W.C.; Weber, T.; Chapman, E.R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 2004, 304, 435–438. [Google Scholar] [CrossRef]
- Vieira, O.V. Rab3a and Rab10 are regulators of lysosome exocytosis and plasma membrane repair. Small GTPases 2018, 9, 349–351. [Google Scholar] [CrossRef]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2019, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef]
- Grimm, C.; Hassan, S.; Wahl-Schott, C.; Biel, M. Role of TRPML and two-pore channels in endolysosomal cation homeostasis. J. Pharmacol. Exp. Ther. 2012, 342, 236–244. [Google Scholar] [CrossRef]
- Turner, C.T.; Fuller, M.; Hopwood, J.J.; Meikle, P.J.; Brooks, D.A. Drug induced exocytosis of glycogen in Pompe disease. Biochem. Biophys. Res. Commun. 2016, 479, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Samie, M.A.; Xu, H. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 2014, 55, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Rammonhan, M.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca2+ Channel Agonists Protects Human Dopaminergic Neurons from α-Synuclein Toxicity. J. Neurosci. 2019, 39, 5760–5772. [Google Scholar] [CrossRef] [PubMed]
- Tancini, B.; Buratta, S.; Sagini, K.; Costanzi, E.; Delo, F.; Urbanelli, L.; Emiliani, C. Insight into the Role of Extracellular Vesicles in Lysosomal Storage Disorders. Genes 2019, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–225. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Möbius, W.; Ohno-Iwashita, Y.; van Donselaar, E.G.; Oorschot, V.M.; Shimada, Y.; Fujimoto, T.; Heijnen, H.F.; Geuze, H.J.; Slot, J.W. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J. Histochem. Cytochem. 2002, 50, 43–55. [Google Scholar] [CrossRef]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Øverbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling pathways in exosomes biogenesis, secretion and fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Casal, E.; Cappello, F.; Carvalho, J.; Colás, E.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef]
- Chairoungdua, A.; Smith, D.L.; Pochard, P.; Hull, M.; Caplan, M.J. Exosome release of β-catenin: A novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 2010, 190, 1079–1091. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Phosphatidic acid in membrane rearrangements. FEBS Lett. 2019, 593, 2428–2451. [Google Scholar] [CrossRef]
- Laulagnier, K.; Grand, D.; Dujardin, A.; Hamdi, S.; Vincent-Schneider, H.; Lankar, D.; Salles, J.P.; Bonnerot, C.; Perret, B.; Record, M. PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett. 2004, 572, 11–14. [Google Scholar] [CrossRef]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef]
- Alonso, R.; Rodríguez, M.C.; Pindado, J.; Merino, E.; Mérida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the secretion of lethal exosomes bearing Fas ligand during activation-induced cell death of T lymphocytes. J. Biol. Chem. 2005, 280, 28439–28450. [Google Scholar] [CrossRef]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ. 2011, 18, 1161–1173. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gelé, P.; Drobeck, H.; Bégard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.; Goebel, C.; Runz, H.; Möbius, W.; Weiss, S.; Feussner, I.; Simons, M.; Schneider, A. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J. Biol. Chem. 2010, 285, 26279–26288. [Google Scholar] [CrossRef] [PubMed]
- Canonico, B.; Cesarini, E.; Salucci, S.; Luchetti, F.; Falcieri, E.; Di Sario, G.; Palma, F.; Papa, S. Defective Autophagy, Mitochondrial Clearance and Lipophagy in Niemann-Pick Type B Lymphocytes. PLoS ONE 2016, 11, e0165780. [Google Scholar] [CrossRef] [PubMed]
- Riva, P.; Battaglia, C.; Venturin, M. Emerging Role of Genetic Alterations Affecting Exosome Biology in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4113. [Google Scholar] [CrossRef] [PubMed]
- Bellingham, S.A.; Guo, B.B.; Coleman, B.M.; Hill, A.F. Exosomes: Vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 2012, 3, 124. [Google Scholar] [CrossRef]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Lai, C.P.; Breakefield, X.O. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front. Physiol. 2012, 3, 228. [Google Scholar] [CrossRef]
- Chen, J.J.; Zhao, B.; Zhao, J.; Li, S. Potential Roles of Exosomal MicroRNAs as Diagnostic Biomarkers and Therapeutic Application in Alzheimer’s Disease. Neural Plast. 2017, 2017, 7027380. [Google Scholar] [CrossRef]
- Vilaça-Faria, H.; Salgado, A.J.; Teixeira, F.G. Mesenchymal Stem Cells-derived Exosomes: A New Possible Therapeutic Strategy for Parkinson’s Disease? Cells 2019, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Machado, E.; White-Gilbertson, S.; van de Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Muallem, S.; Kim, S.H.; Kwon, K.B.; Kim, M.S. Exosomal release through TRPML1-mediated lysosomal exocytosis is required for adipogenesis. Biochem. Biophys. Res. Commun. 2019, 51, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhu, Y.L.; Hu, F.H.; Wang, Y.Y.; Huang, N.P.; Xiao, Z.D. Dynamics of exosome internalization and trafficking. J. Cell Physiol. 2013, 228, 1487–1495. [Google Scholar] [CrossRef]
- Chiba, M.; Watanabe, N.; Watanabe, M.; Sakamoto, M.; Sato, A.; Fujisaki, M.; Kubota, S.; Monzen, S.; Maruyama, A.; Nanashima, N.; et al. Exosomes derived from SW480 colorectal cancer cells promote cell migration in HepG2 hepatocellular cancer cells via the mitogen-activated protein kinase pathway. Int. J. Oncol. 2016, 48, 305–312. [Google Scholar] [CrossRef][Green Version]
- Harnett, M.M.; Pineda, M.A.; Latré de Laté, P.; Eason, R.J.; Besteiro, S.; Harnett, W.; Langsley, G. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed. J. 2017, 40, 9–22. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Cadwell, K.; Debnath, J. Beyond self-eating: The control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 2018, 217, 813–822. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, M.; Li, X.; Liu, J.; Li, H. Origin of the Autophagosome membrane in mammals. BioMed Res. Int. 2018, 2018, 1012789. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Eskelinen, E.L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes. Wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Development of Research into Autophagic Lysosome Reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Olsvik, H.L.; Svenning, S.; Abudu, Y.P.; Brech, A.; Stenmark, H.; Johansen, T.; Mejlvang, J. Endosomal microautophagy is an integrated part of the autophagic response to amino acid starvation. Autophagy 2019, 15, 182–183. [Google Scholar] [CrossRef]
- Gudbergsson, J.M.; Johnsen, K.B. Exosomes and autophagy: Rekindling the vesicular waste hypothesis. J. Cell Commun. Signal. 2019, 13, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Claude-Taupin, A.; Bissa, B.; Jia, J.; Gu, Y.; Deretic, V. Role of autophagy in IL-1β export and release from cells. Semin. Cell Dev. Biol. 2018, 83, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Cozzolino, F.; Talio, M.; Sitia, R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990, 9, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy 2020, 1–24. [Google Scholar] [CrossRef]
- Nüchel, J.; Ghatak, S.; Zuk, A.V.; Illerhaus, A.; Mörgelin, M.; Schönborn, K.; Blumbach, K.; Wickström, S.A.; Krieg, T.; Sengle, G.; et al. TGFB1 is secreted through an unconventional pathway dependent on the autophagic machinery and cytoskeletal regulators. Autophagy 2018, 14, 465–486. [Google Scholar]
- Noh, S.H.; Gee, H.Y.; Kim, Y.; Piao, H.; Kim, J.; Kang, C.M.; Lee, G.; Mook-Jung, I.; Lee, Y.; Cho, J.W.; et al. Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy 2018, 14, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.O.; Gregory, S.; Hu, H.; Chao, Y.; Sepúlveda, V.E.; He, Y.; Li-Kroeger, D.; Goldman, W.E.; Bellen, H.J.; Venkatachalam, K. Lysosomal Degradation Is Required for Sustained Phagocytosis of Bacteria by Macrophages. Cell Host Microbe 2017, 21, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Romao, S.; Münz, C. LC3-associated phagocytosis. Autophagy 2014, 10, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018, 37, e97840. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Subbarao Malireddi, R.K.; Lu, Q.; Dias Cunha, L.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis (LAP) reveals distinct roles for Rubicon, NOX2, and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef]
- Wong, S.W.; Sil, P.; Martinez, J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J. 2018, 285, 1379–1388. [Google Scholar] [CrossRef]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef]
- Florey, O.; Overholtzer, M. Macropinocytosis and autophagy crosstalk in nutrient scavenging. Philos. Trans. R. Soc. B 2019, 374, 20180154. [Google Scholar] [CrossRef]
- Gerstenmaier, L.; Pilla, R.; Herrmann, L.; Herrmann, H.; Prado, M.; Villafano, G.J.; Kolonko, M.; Reimer, R.; Soldati, T.; King, J.S.; et al. The autophagic machinery ensures nonlytic transmission of mycobacteria. Proc. Natl. Acad. Sci. USA 2015, 112, E687–E692. [Google Scholar] [CrossRef]
- Starr, T.; Child, R.; Wehrly, T.D.; Hansen, B.; Hwang, S.; López-Otin, C.; Virgin, H.W.; Celli, J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012, 11, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Nakai, M.; Iwamaru, Y.; Sugama, S.; Tsukimoto, M.; Fujita, M.; Wei, J.; Sekigawa, A.; Sato, M.; Kojima, S.; et al. The Activation of P2X7 Receptor Impairs Lysosomal Functions and Stimulates the Release of Autophagolysosomes in Microglial Cells. J. Immunol. 2009, 182, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Mutsafi, Y.; Altan-Bonnet, N. Enterovirus Transmission by Secretory Autophagy. Viruses 2018, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Li, Z.L.; Yuan, S. The Role of Secretory Autophagy in Zika Virus Transfer through the Placental Barrier. Front. Cell Infect. Microbiol. 2017, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef]
- Nolte, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Ann. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Klionsky, D.J. Secretory autophagy holds the key to lysozyme secretion during bacterial infection of the intestine. Autophagy 2018, 14, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef] [PubMed]
- Ejlerskov, P.; Rasmussen, I.; Nielsen, T.T.; Bergström, A.L.; Tohyama, Y.; Jensen, P.H.; Vilhardt, F. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosomelysosome fusion. J. Biol. Chem. 2013, 288, 17313–17335. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef]
- Yuyama, K.; Igarashi, Y. Exosomes as Carriers of Alzheimer’s Amyloid-ß. Front. Neurosci. 2017, 11, 229. [Google Scholar] [CrossRef]
- Tian, Y.; Bustos, V.; Flajolet, M.; Greengard, P. A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011, 25, 1934–1942. [Google Scholar] [CrossRef]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef]
- Kang, S.; Son, S.M.; Baik, S.H.; Yang, J.; Mook-Jung, I. Autophagy-Mediated Secretory Pathway is Responsible for Both Normal and Pathological Tau in Neurons. J. Alzheimers Dis. 2019, 70, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Cha, M.Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.S.; Park, S.A.; Mook-Junga, I. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Manjithaya, R.; Subramani, S. Role of autophagy in unconventional protein secretion. Autophagy 2010, 6, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Kinseth, M.A.; Anjard, C.; Fuller, D.; Guizzunti, G.; Loomis, W.F.; Malhotra, V. The Golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell 2007, 130, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Jiang, S.; Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012, 22, 397–406. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Gordon, P.B.; Seglen, P.O. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 1988, 151, 40–47. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Fader, C.M.; Sánchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Bader, C.A.; Shandala, T.; Ng, Y.S.; Johnson, I.R.; Brooks, D.A. Atg9 is required for intraluminal vesicles in amphisomes and autolysosomes. Biol. Open 2015, 4, 1345–1355. [Google Scholar] [CrossRef]
- Chen, Y.D.; Fang, Y.T.; Cheng, Y.L.; Lin, C.F.; Hsu, L.J.; Wang, S.Y.; Anderson, R.; Chang, C.P.; Lin, Y.S. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-γ-stimulated lung epithelial cells. Sci. Rep. 2017, 7, 5676. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R.E.; Kupzig, S.; Cogan, N.; Mankelow, T.J.; Betin, V.M.; Trakarnsanga, K.; Massey, E.J.; Lane, J.D.; Parsons, S.F.; Anstee, D.J. Maturing reticulocytes internalize plasma membrane in glycophorin A-containing vesicles that fuse with autophagosomes before exocytosis. Blood 2012, 119, 6296–6306. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.L.; Saitoh, T.; Akira, S.; Seglen, P.O.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chitiprolu, M.; Roncevic, L.; Javalet, C.; Hemming, F.J.; Trung, M.T.; Meng, L.; Latreille, E.; Tanese de Souza, C.; McCulloch, D.; et al. Atg5 Disassociates the V1V0-ATPase to Promote Exosome Production and Tumor Metastasis Independent of Canonical Macroautophagy. Dev. Cell 2017, 43, 716–730. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Murrow, L.; Malhotra, R.; Debnath, J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 2015, 17, 300–310. [Google Scholar] [CrossRef]
- Papandreou, M.E.; Tavernarakis, N. Autophagy and the endo/exosomal pathways in health and disease. Biotechnol. J. 2017, 12, 1600175. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. https://doi.org/10.3390/ijms21072576
Buratta S, Tancini B, Sagini K, Delo F, Chiaradia E, Urbanelli L, Emiliani C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. International Journal of Molecular Sciences. 2020; 21(7):2576. https://doi.org/10.3390/ijms21072576
Chicago/Turabian StyleBuratta, Sandra, Brunella Tancini, Krizia Sagini, Federica Delo, Elisabetta Chiaradia, Lorena Urbanelli, and Carla Emiliani. 2020. "Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular" International Journal of Molecular Sciences 21, no. 7: 2576. https://doi.org/10.3390/ijms21072576
APA StyleBuratta, S., Tancini, B., Sagini, K., Delo, F., Chiaradia, E., Urbanelli, L., & Emiliani, C. (2020). Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. International Journal of Molecular Sciences, 21(7), 2576. https://doi.org/10.3390/ijms21072576