Mitochondrial Damage and Necroptosis in Aging Cochlea

 , and
, and 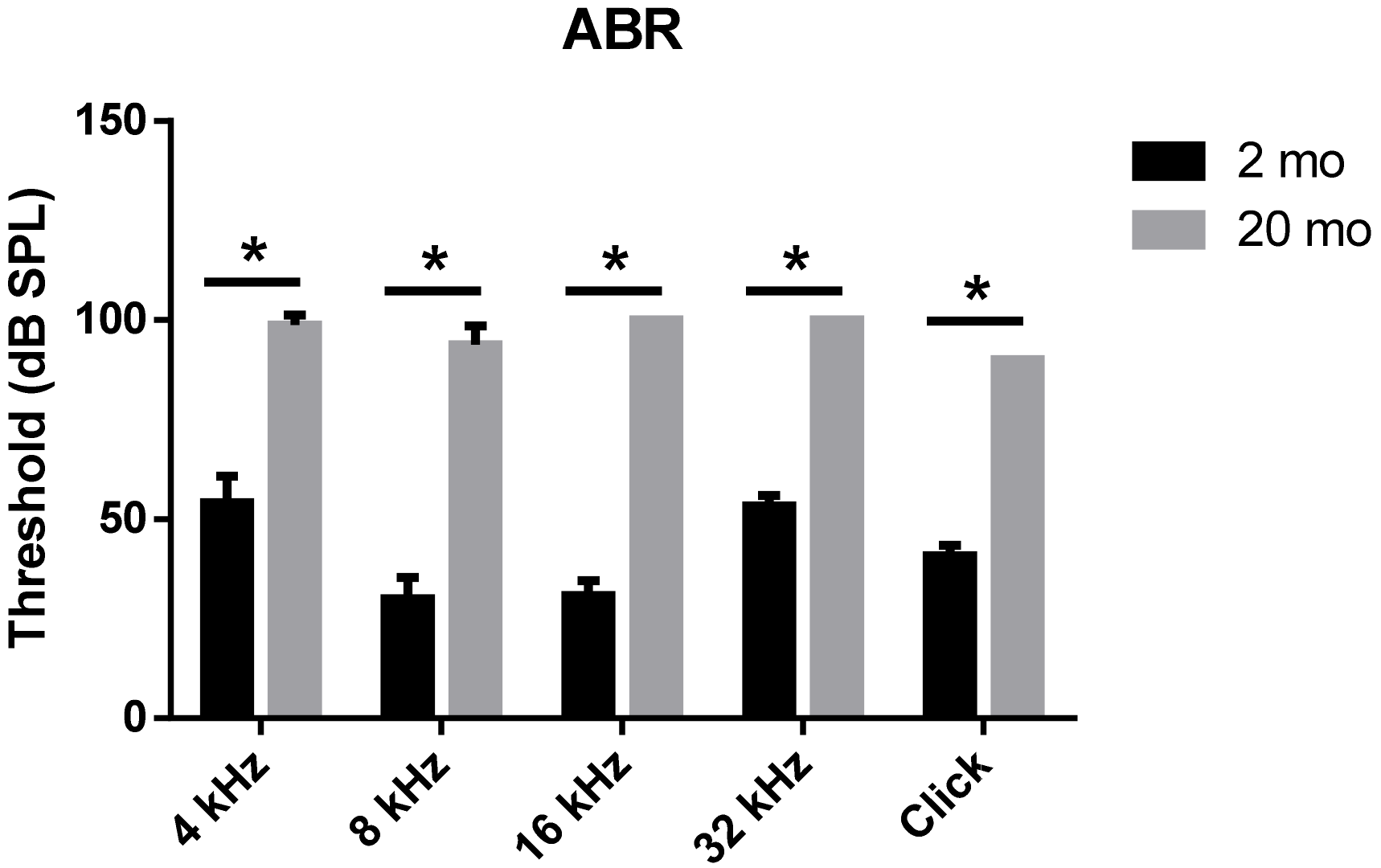{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Design
2.2. Auditory Brainstem Response (ABR)
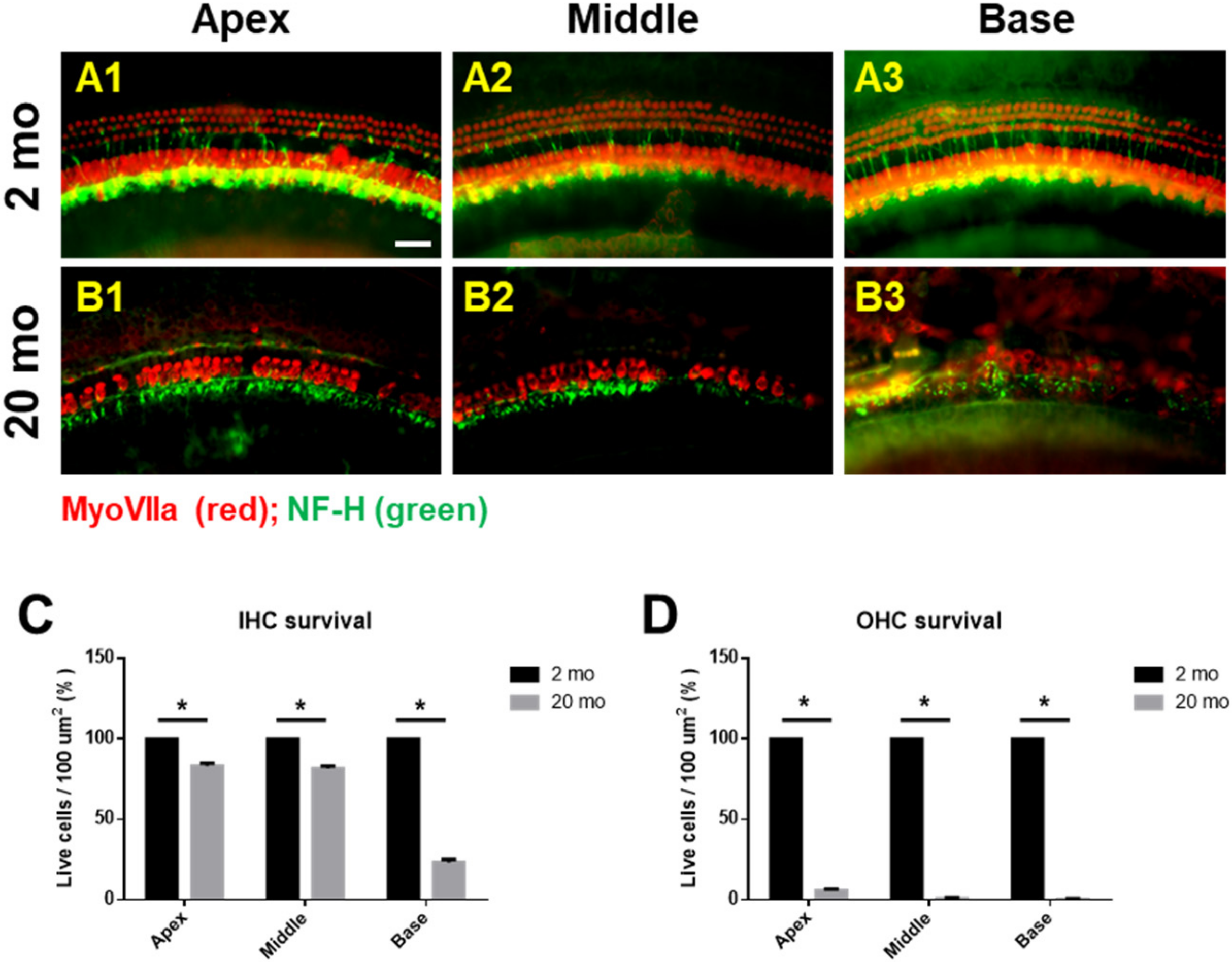
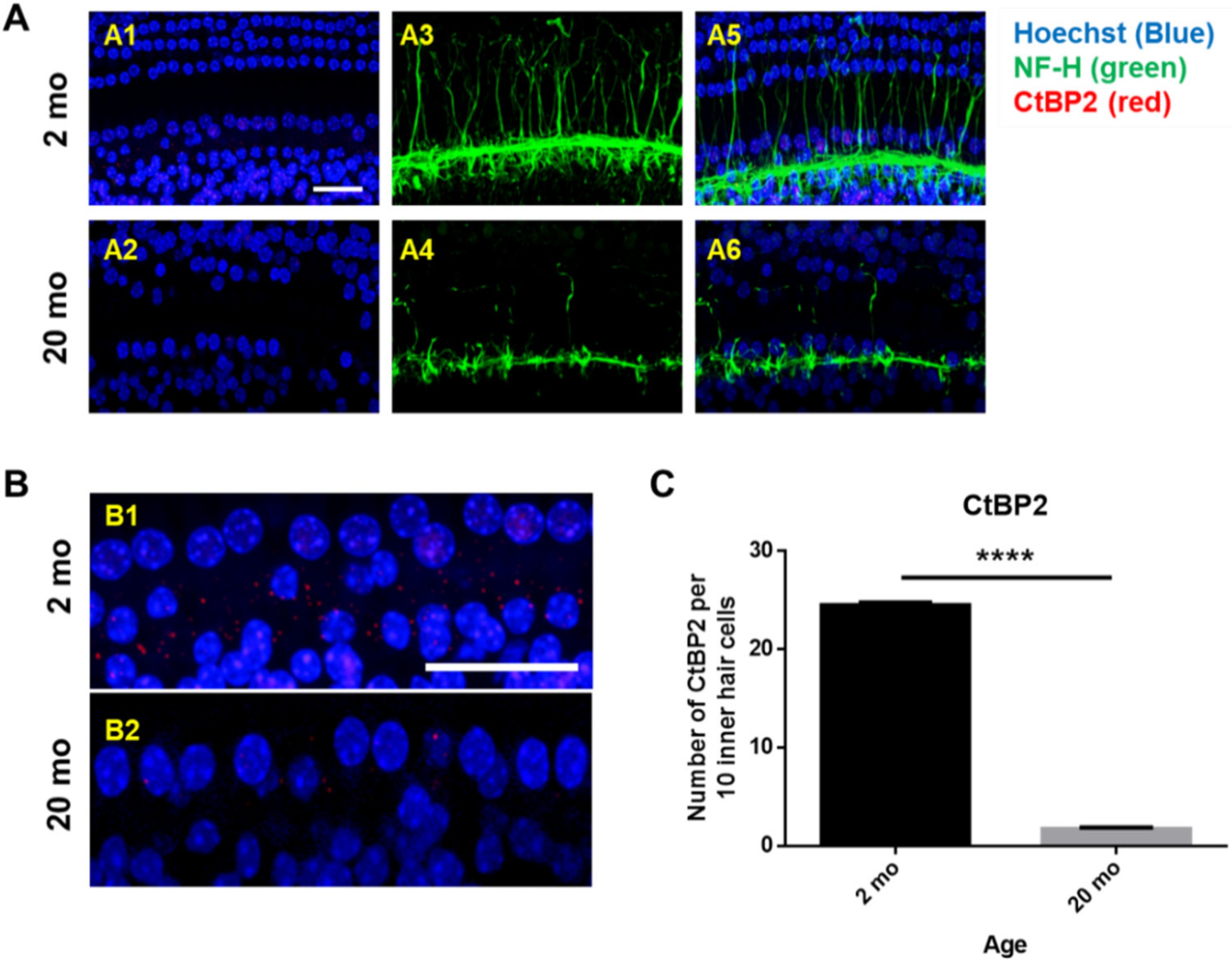
2.3. Tissue Preparation and Immunofluorescence
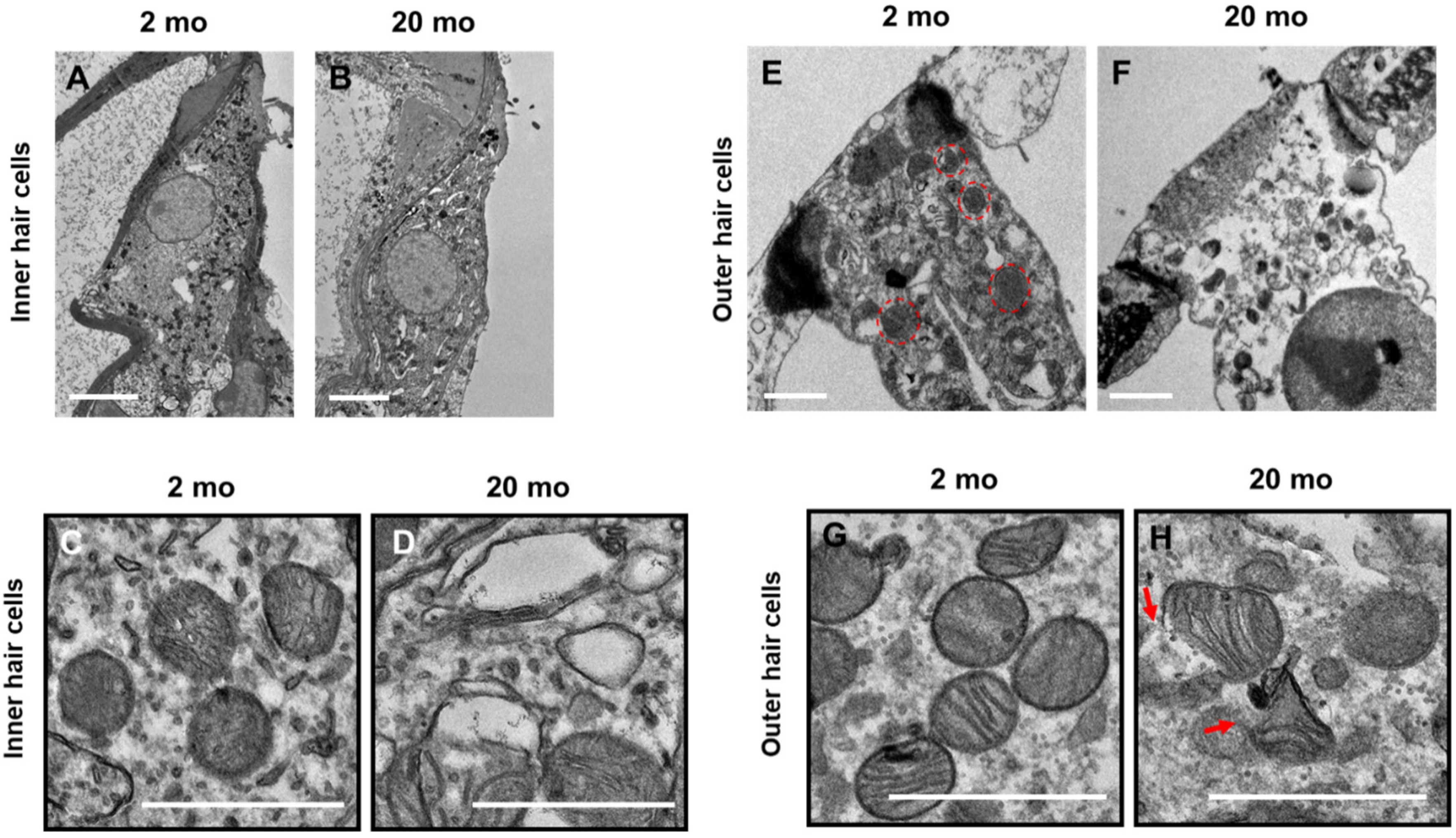
2.4. Transmission Electron Microscopy (TEM)
2.5. Measurement of Cochlear Blood Flow
2.6. Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
2.7. Image Processing and Statistical Analysis
3. Results
3.1. Auditory Brainstem Response Thresholds and Histopathologies of Young and Aging Cochleae
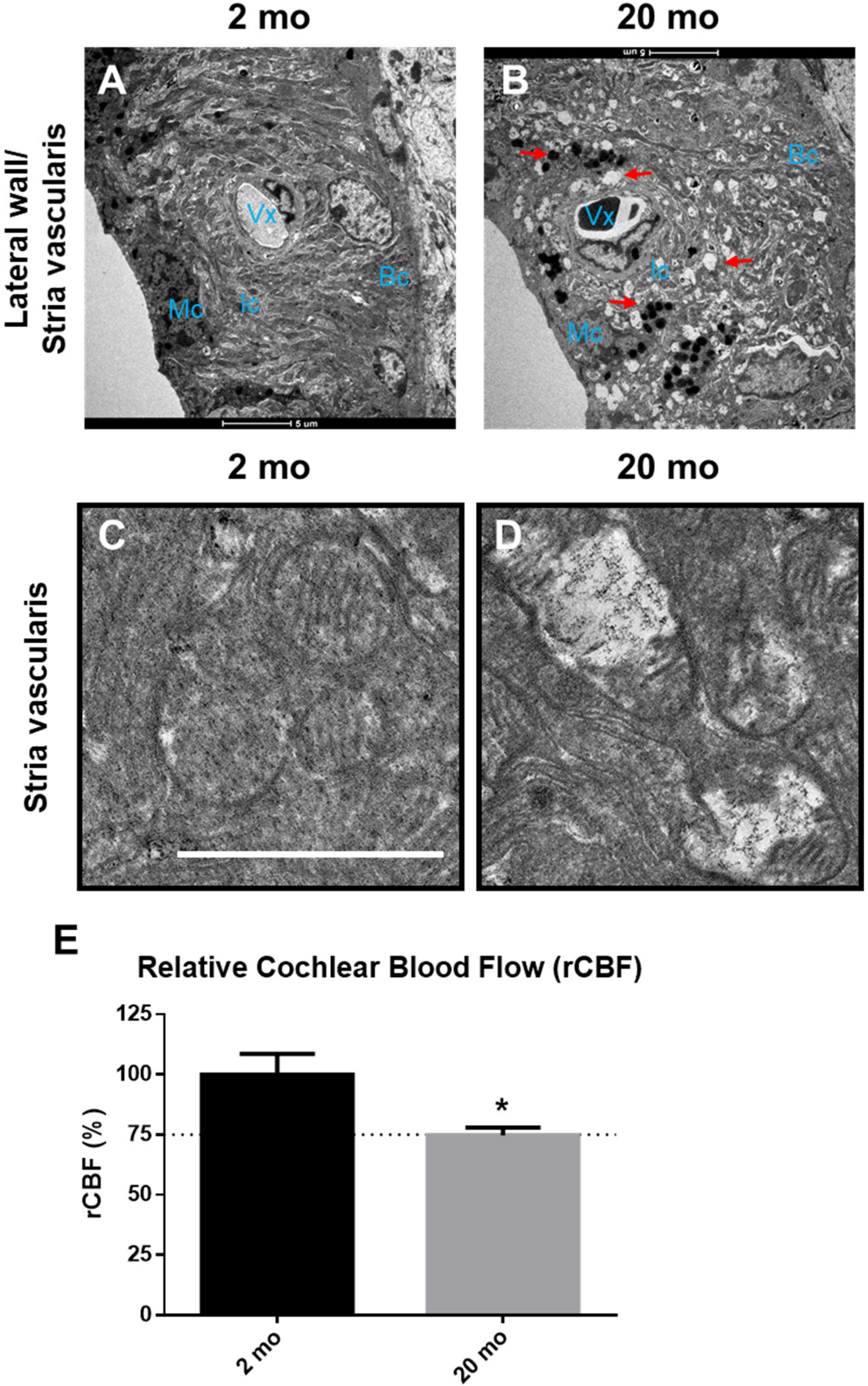
3.2. Cochlear Microcirculation and Morphologies
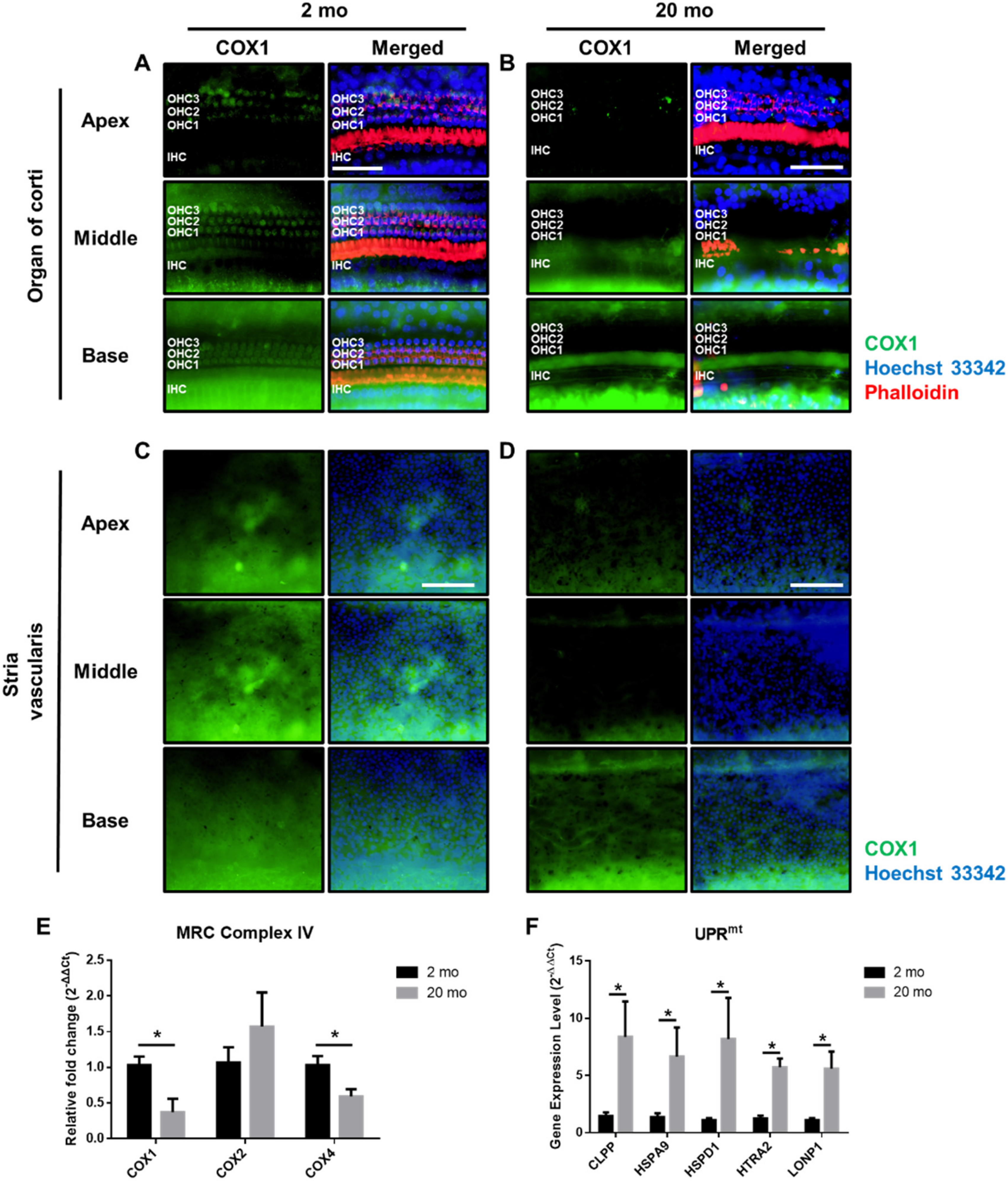
3.3. Mitochondrial Damage and Stress Responses
3.4. Aging Cochleae Exhibit Necroptosis and An Inflammatory Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABR | Auditory brainstem response |
| ARHL | Age-related hearing loss |
| Bak | BCL2-antagonist/killer 1 |
| Bc | Basal cells |
| Cdh23 | Cadeherin 23 |
| COX | Cytochrome c oxidase |
| COX1 | Cytochrome C oxidase I |
| COX4 | Cytochrome C oxidase Ⅳ |
| CLPP | Caseinolytic mitochondrial matrix peptidase proteolytic subunit |
| DNM1L | Dynamin-1-like protein |
| Drp1 | Dynamin-related protein 1 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HSPA9 | Heat shock 70-kDa protein |
| HSPD1 | Heat shock 60-kDa protein 1 |
| HTRA2 | HtrA serine peptidase 2 |
| Ic | Intermediate cells |
| IHC | Inner hair cells |
| LPNP1 | Mitochondrial ion peptidase 1 |
| Mc | Marginal cells |
| MLKL | Mixed-lineage kinase domain-like |
| Nec1 | Necrostatin-1 |
| OHCs | Outer hair cells |
| PBS | Phosphate-buffered saline |
| qRT-PCR | Quantitative real time polymerase chain reaction |
| RHIM | RIP homology interaction motif |
| RIP | Receptor-interacting protein |
| ROS | Reactive oxygen species |
| SGNs | Spiral ganglion neurons |
| TEM | Transmission Electron Microscopy |
| UPR | Unfolded protein response |
| UPRmt | Mitochondrial unfolded protein response |
References
- Bowl, M.R.; Dawson, S.J. Age-related hearing loss. Cold Spring Harb. Perspect. Med. 2019, 9, a033217. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, T.; Lin, F.R.; Someya, S.; Kashio, A.; Sakamoto, T.; Kondo, K. Current concepts in age-related hearing loss: Epidemiology and mechanistic pathways. Hear. Res. 2013, 303, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, N.; Lin, F.R.; Someya, S.; Kashio, A.; Sakamoto, T.; Kondo, K. Epidemiology of age related hearing loss: A review. Hear. Res. 2015, 13, 77–81. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Addressing the Rising Prevalence of Hearing Loss; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Fujimoto, C.; Yamasoba, T. Oxidative stresses and mitochondrial dysfunction in age-related hearing loss. Oxidative Med. Cell. Longev. 2014, 2014, 582849. [Google Scholar] [CrossRef]
- Henry, K.R.; Chole, R.A. Genotypic differences in behavioral, physiological and anatomical expressions of age-related hearing loss in the laboratory mouse. Audiol. Off. Organ Int. Soc. Audiol. 1980, 19, 369–383. [Google Scholar] [CrossRef]
- Hequembourg, S.; Liberman, M.C. Spiral ligament pathology: A major aspect of age-related cochlear degeneration in C57BL/6 mice. J. Assoc. Res. Otolaryngol. 2001, 2, 118–129. [Google Scholar] [CrossRef]
- Mikaelian, D.O. Development and degeneration of hearing in the C57/b16 mouse: Relation of electrophysiologic responses from the round window and cochlear nucleus to cochlear anatomy and behavioral responses. Laryngoscope 1979, 89, 1–15. [Google Scholar] [CrossRef]
- Han, F.; Wang, O.; Cai, Q. Anti-apoptotic treatment in mouse models of age-related hearing loss. J. Otol. 2016, 11, 7–12. [Google Scholar] [CrossRef]
- Someya, S.; Xu, J.; Kondo, K.; Ding, D.; Salvi, R.J.; Yamasoba, T.; Rabinovitch, P.S.; Weindruch, R.; Leeuwenburgh, C.; Tanokura, M.; et al. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 19432–19437. [Google Scholar] [CrossRef]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef]
- Fujimoto, C.; Yamasoba, T. Mitochondria-Targeted Antioxidants for Treatment of Hearing Loss: A Systematic Review. Antioxidants (Basel) 2019, 8, 109. [Google Scholar] [CrossRef]
- Sun, H.Y.; Hu, Y.J.; Zhao, X.Y.; Zhong, Y.; Zeng, L.L.; Chen, X.B.; Yuan, J.; Wu, J.; Sun, Y.; Kong, W.; et al. Age-related changes in mitochondrial antioxidant enzyme Trx2 and TXNIP-Trx2-ASK1 signal pathways in the auditory cortex of a mimetic aging rat model: Changes to Trx2 in the auditory cortex. FEBS J. 2015, 282, 2758–2774. [Google Scholar] [CrossRef]
- Ding, D.; Jiang, H.; Chen, G.; Longo-Guess, C.; Muthaiah, V.P.K.; Tian, C.; Sheppard, A.; Salvi, R.; Johnson, K.R. N-acetyl-cysteine prevents age-related hearing loss and the progressive loss of inner hair cells in gamma-glutamyl transferase 1 deficient mice. Aging 2016, 8, 730–750. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Kim, M.-J.; Han, C.; Park, H.-J. Loss of IDH2 Accelerates Age-related Hearing Loss in Male Mice. Sci. Rep. 2018, 8, 5039. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Prolla, T.A. Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech. Ageing Dev. 2010, 131, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef]
- Kurabi, A.; Keithley, E.M.; Housley, G.D.; Ryan, A.F.; Wong, A.C. Cellular mechanisms of noise-induced hearing loss. Hear. Res. 2017, 349, 129–137. [Google Scholar] [CrossRef]
- Lyu, A.R.; Kim, D.H.; Lee, S.H.; Shin, D.S.; Shin, S.A.; Park, Y.H. Effects of dexamethasone on intracochlear inflammation and residual hearing after cochleostomy: A comparison of administration routes. BioMed Res. Int. 2018, 13, e0195230. [Google Scholar] [CrossRef]
- Lee, S.H.; Lyu, A.R.; Shin, S.A.; Jeong, S.H.; Lee, S.A.; Park, M.J.; Park, Y.H. Cochlear Glucocorticoid Receptor and Serum Corticosterone Expression in a Rodent Model of Noise-induced Hearing Loss: Comparison of Timing of Dexamethasone Administration. Sci. Rep. 2019, 9, 12646. [Google Scholar] [CrossRef]
- Shin, S.A.; Lyu, A.-R.; Jeong, S.-H.; Kim, T.H.; Park, M.J.; Park, Y.-H. Acoustic Trauma Modulates Cochlear Blood Flow and Vasoactive Factors in a Rodent Model of Noise-Induced Hearing Loss. Int. J. Mol. Sci. 2019, 20, 5316. [Google Scholar] [CrossRef]
- Han, W.K.; Kim, E.H.; Shin, S.A.; Shin, D.S.; Kim, B.J.; Lyu, A.R.; Park, Y.H. Susceptibility of Diabetic Mice to Noise Trauma. Biomed. Res. Int. 2018, 2018, 7601232. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C T method. Nat. Protoc. 2008, 3, 1101. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yang, Y.; Hu, Y.; Sun, Y.; Du, Z.; Xie, Z.; Zhou, T.; Kong, W. Age-related decrease in the mitochondrial sirtuin deacetylase Sirt3 expression associated with ROS accumulation in the auditory cortex of the mimetic aging rat model. PLoS ONE 2014, 9, e88019. [Google Scholar] [CrossRef] [PubMed]
- Olmos, P.R.; Borzone, G.R.; Olmos, J.P.; Diez, A.; Santos, J.L.; Serrano, V.; Cataldo, L.R.; Anabalón, J.L.; Correa, C.H. Mitochondrial diabetes and deafness: Possible dysfunction of strial marginal cells of the inner ear. J. Otolaryngol. 2011, 40, 93–103. [Google Scholar]
- Fischel-Ghodsian, N.; Kopke, R.D.; Ge, X. Mitochondrial dysfunction in hearing loss. Mitochondrion 2004, 4, 675–694. [Google Scholar] [CrossRef]
- Han, C.; Someya, S. Mouse models of age-related mitochondrial neurosensory hearing loss. Mol. Cell Neurosci. 2013, 55, 95–100. [Google Scholar] [CrossRef]
- Fontanesi, F.; Soto, I.C.; Barrientos, A. Cytochrome c oxidase biogenesis: New levels of regulation. IUBMB Life 2008, 60, 557–568. [Google Scholar] [CrossRef]
- Park, M.J.; Aja, S.; Li, Q.; Degano, A.L.; Penati, J.; Zhuo, J.; Roe, C.R.; Ronnett, G.V. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE 2014, 9, e109527. [Google Scholar] [CrossRef]
- Shao, L.; Yu, S.; Ji, W.; Li, H.; Gao, Y. The Contribution of Necroptosis in Neurodegenerative Diseases. Neurochem. Res. 2017, 42, 2117–2126. [Google Scholar] [CrossRef]
- Oliveira, S.R.; Amaral, J.D.; Rodrigues, C.M.P. Mechanism and disease implications of necroptosis and neuronal inflammation. Cell Death Dis. 2018, 9, 903. [Google Scholar] [CrossRef]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef]
- Kane, K.L.; Longo-Guess, C.M.; Gagnon, L.H.; Ding, D.; Salvi, R.J.; Johnson, K.R. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear. Res. 2012, 283, 80–88. [Google Scholar] [CrossRef]
- Zheng, Q.Y.; Johnson, K.R.; Erway, L.C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear. Res. 1999, 130, 94–107. [Google Scholar] [CrossRef]
- Noben-Trauth, K.; Johnson, K.R. Inheritance patterns of progressive hearing loss in laboratory strains of mice. Brain Res. 2009, 1277, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Borg, E. Age-related loss of auditory sensitivity in two mouse genotypes. Acta Otolaryngol. 1991, 111, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Erway, L.C.; Cook, S.A.; Willott, J.F.; Zheng, Q.Y. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear. Res. 1997, 114, 83–92. [Google Scholar] [CrossRef]
- Frisina, R.D.; Singh, A.; Bak, M.; Bozorg, S.; Seth, R.; Zhu, X. F1 (CBA× C57) mice show superior hearing in old age relative to their parental strains: Hybrid vigor or a new animal model for “Golden Ears”? Neurobiol. Aging 2011, 32, 1716–1724. [Google Scholar] [CrossRef]
- van de Ven, R.A.H.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol. Med. 2017, 23, 320–331. [Google Scholar] [CrossRef]
- Kwon, D.N.; Park, W.J.; Choi, Y.J.; Gurunathan, S.; Kim, J.H. Oxidative stress and ROS metabolism via down-regulation of sirtuin 3 expression in Cmah-null mice affect hearing loss. Aging 2015, 7, 579–594. [Google Scholar] [CrossRef]
- Wang, W.; Suna, Y.; Chen, S.; Zhou, X.; Wu, X.; Kong, W.; Kong, W. Impaired unfolded protein response in the degeneration of cochlea cells in a mouse model of age-related hearing loss. Exp. Gerontol. 2015, 70, 61–70. [Google Scholar] [CrossRef]
- Suzukawa, K.; Tomlin, J.; Pak, K.; Chavez, E.; Kurabi, A.; Baird, A.; Wasserman, S.I.; Ryan, A.F. A mouse model of otitis media identifies HB-EGF as a mediator of inflammation-induced mucosal proliferation. PLoS ONE 2014, 9, e102739. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Zhu, Y.; Walsh, T.; Xie, D.; Yuan, H.; Sirmaci, A.; Fujikawa, T.; Wong, A.C.Y.; Loh, T.L.; Du, L.; et al. Mutation of the ATP-gated P2X(2) receptor leads to progressive hearing loss and increased susceptibility to noise. Proc. Natl. Acad. Sci. USA 2013, 110, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Sakamoto, T.; Kashio, A.; Shimizu, T.; Yamasoba, T. Age-related hearing loss in Mn-SOD heterozygous knockout mice. Oxidative Med. Cell. Longev. 2013, 2013, 325702. [Google Scholar] [CrossRef] [PubMed]
- Lauer, A.M.; Fuchs, P.A.; Ryugo, D.K.; Francis, H.W. Efferent synapses return to inner hair cells in the aging cochlea. Neurobiol. Aging 2012, 33, 2892–2902. [Google Scholar] [CrossRef]
- Coling, D.E.; Yu, K.C.; Somand, D.; Satar, B.; Bai, U.; Huang, T.T.; Seidman, M.D.; Epstein, C.J.; Mhatre, A.N.; Lalwani, A.K. Effect of SOD1 overexpression on age- and noise-related hearing loss. Free Radic. Biol. Med. 2003, 34, 873–880. [Google Scholar] [CrossRef]
- Keithley, E.M.; Erkman, L.; Bennett, T.; Lou, L.; Ryan, A.F. Effects of a hair cell transcription factor, Brn-3.1, gene deletion on homozygous and heterozygous mouse cochleas in adulthood and aging. Hear. Res. 1999, 134, 71–76. [Google Scholar] [CrossRef]
- Briner, W.; Willott, J.F. Ultrastructural features of neurons in the C57BL/6J mouse anteroventral cochlear nucleus: Young mice versus old mice with chronic presbycusis. Neurobiol. Aging 1989, 10, 295–303. [Google Scholar] [CrossRef]
- Choi, M.J.; Kang, H.; Lee, Y.Y.; Choo, O.; Jang, J.H.; Park, S.; Moon, J.; Choi, S.J.; Choung, Y. Cisplatin-Induced Ototoxicity in Rats Is Driven by RIP3-Dependent Necroptosis. Cells 2019, 8, 409. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Ruhl, D.; Du, T.T.; Wagner, E.L.; Choi, J.H. Necroptosis and Apoptosis Contribute to Cisplatin and Aminoglycoside Ototoxicity. J. Neurosci. 2019, 39, 2951–2964. [Google Scholar] [CrossRef]
- Zheng, H.W.; Chen, J.; Sha, S.H. Receptor-interacting protein kinases modulate noise-induced sensory hair cell death. Cell Death Dis. 2014, 5, e1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Ding, Z.J.; Yue, B.; Zhang, P.Z.; Chen, X.D.; Chen, X.; Chen, J.; Chen, F.Q.; Chen, Y.; et al. The role of RIP3 mediated necroptosis in ouabain-induced spiral ganglion neurons injuries. Neurosci. Lett. 2014, 578, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Lee, B.D.; Chae, S.W.; Chi, J.; Kwon, S.K.; Song, J.J. Protective effect of NecroX, a novel necroptosis inhibitor, on gentamicin-induced ototoxicity. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Molnar, M.; Garnham, C.; Benav, H.; Rask-Andersen, H. Macrophages in the Human Cochlea: Saviors or Predators-A Study Using Super-Resolution Immunohistochemistry. Front. Immunol. 2018, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, J.T.; Nadol, J.B., Jr.; McKenna, M.J. Anti CD163+, Iba1+, and CD68+ Cells in the Adult Human Inner Ear: Normal Distribution of an Unappreciated Class of Macrophages/Microglia and Implications for Inflammatory Otopathology in Humans. Otol. Neurotol. 2016, 37, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Z.; Li, L.; Zhong, C.Q.; Zheng, X.; Wu, X.; Zhang, Y.; Ma, H.; Huang, D.; Li, W.; et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 2013, 288, 16247–16261. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, F.; Chen, Y.; Luo, J.; Liu, S.; Zhang, B.; Ye, Z.; Wang, W.; Liang, X.; Shi, W. Necrostatin-1 attenuates ischemia injury induced cell death in rat tubular cell line NRK-52E through decreased Drp1 expression. Int. J. Mol. Sci. 2013, 14, 24742–24754. [Google Scholar] [CrossRef]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, A.-R.; Kim, T.H.; Park, S.J.; Shin, S.-A.; Jeong, S.-H.; Yu, Y.; Huh, Y.H.; Je, A.R.; Park, M.J.; Park, Y.-H. Mitochondrial Damage and Necroptosis in Aging Cochlea. Int. J. Mol. Sci. 2020, 21, 2505. https://doi.org/10.3390/ijms21072505
Lyu A-R, Kim TH, Park SJ, Shin S-A, Jeong S-H, Yu Y, Huh YH, Je AR, Park MJ, Park Y-H. Mitochondrial Damage and Necroptosis in Aging Cochlea. International Journal of Molecular Sciences. 2020; 21(7):2505. https://doi.org/10.3390/ijms21072505
Chicago/Turabian StyleLyu, Ah-Ra, Tae Hwan Kim, Sung Jae Park, Sun-Ae Shin, Seong-Hun Jeong, Yang Yu, Yang Hoon Huh, A Reum Je, Min Jung Park, and Yong-Ho Park. 2020. "Mitochondrial Damage and Necroptosis in Aging Cochlea" International Journal of Molecular Sciences 21, no. 7: 2505. https://doi.org/10.3390/ijms21072505
APA StyleLyu, A.-R., Kim, T. H., Park, S. J., Shin, S.-A., Jeong, S.-H., Yu, Y., Huh, Y. H., Je, A. R., Park, M. J., & Park, Y.-H. (2020). Mitochondrial Damage and Necroptosis in Aging Cochlea. International Journal of Molecular Sciences, 21(7), 2505. https://doi.org/10.3390/ijms21072505