Targeted Integration of Inducible Caspase-9 in Human iPSCs Allows Efficient in vitro Clearance of iPSCs and iPSC-Macrophages

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
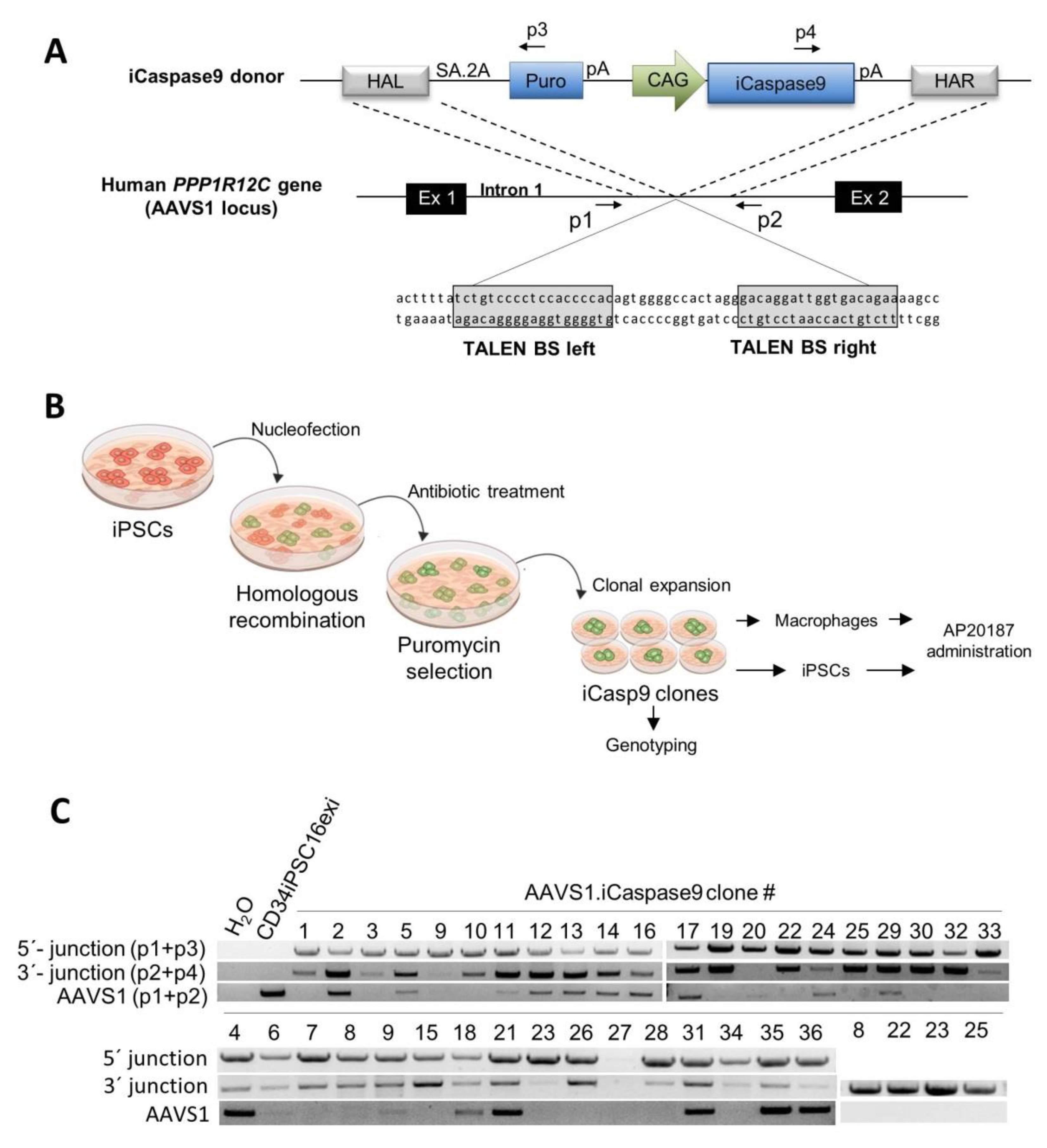
2.1. Generation of iCasp9 Expressing Human iPSC Lines
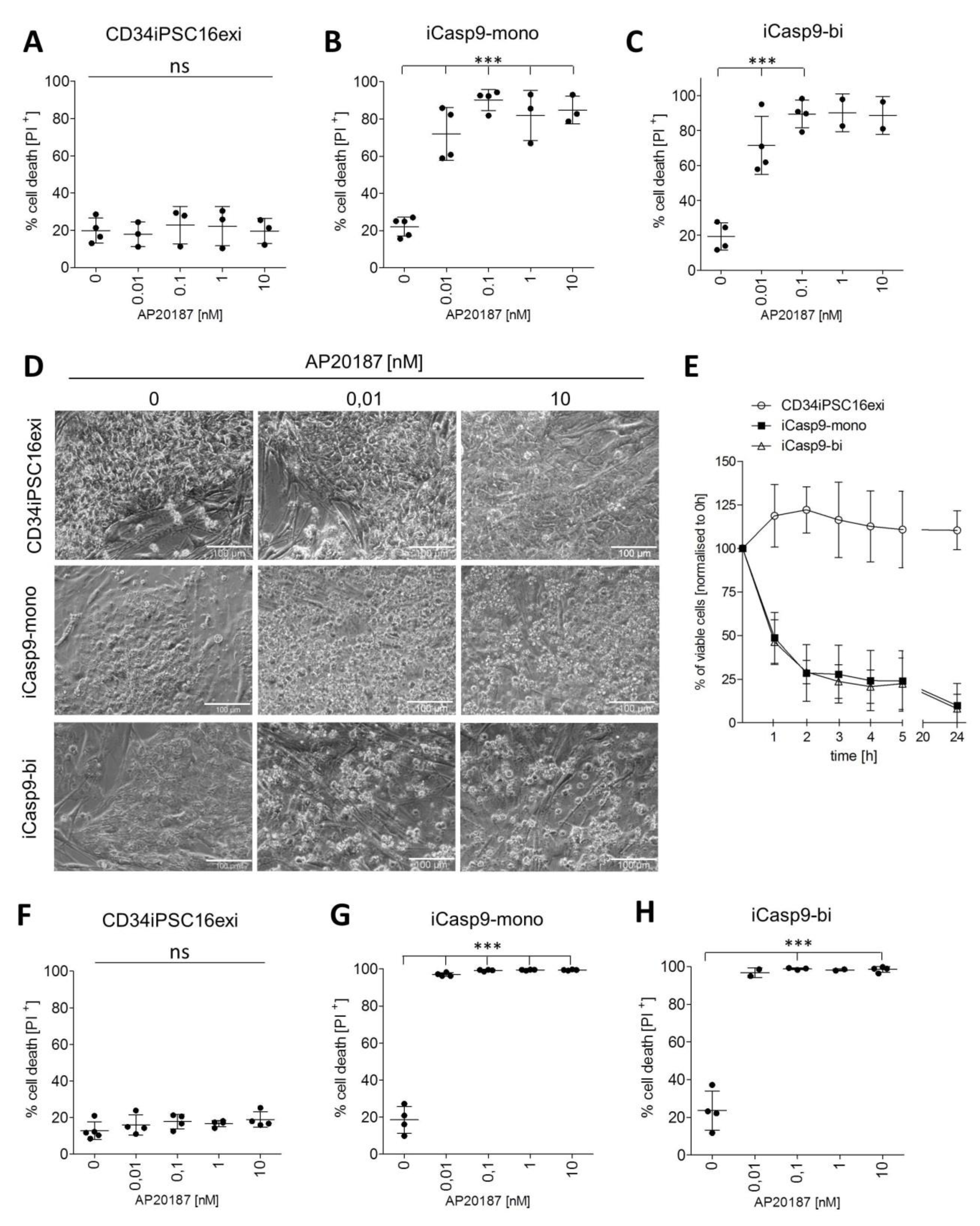
2.2. Effective Killing of iCasp9-iPSCs
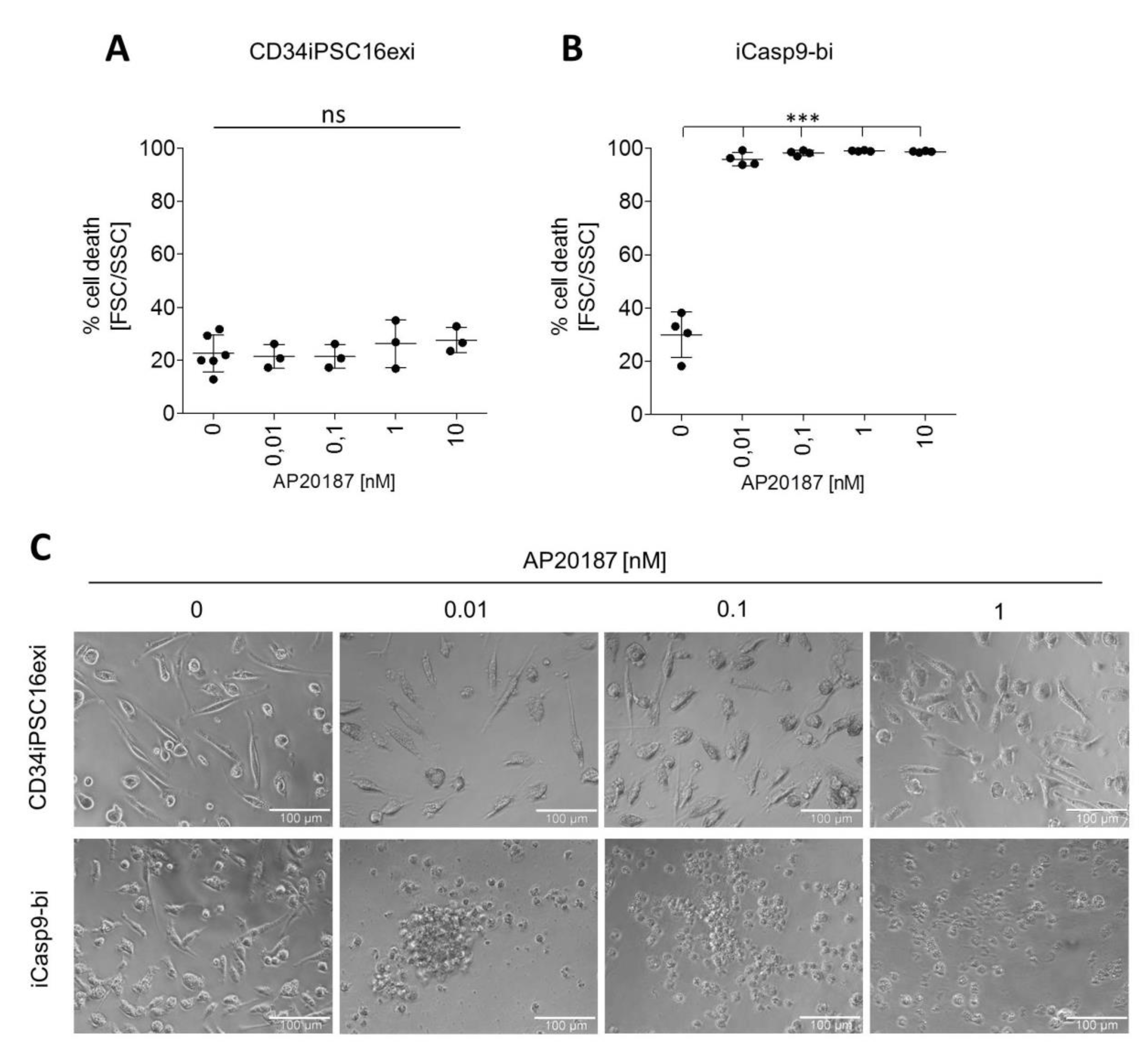
2.3. Macrophage Differentiation from iCasp9-iPSCs
2.4. Induction of Killing in iMΦ
3. Discussion
4. Materials and Methods
4.1. Plasmids and Donors
4.2. Cell Culture
4.3. Nucleofection and Gene Editing of Human iPSC
4.4. Genotyping of Gene-Edited iPSCs
4.5. Real-Time Quantitative Reverse Transcription PCR Analysis
4.6. Alkaline Phosphatase Staining
4.7. Hematopoietic Differentiation of Human iPSCs Toward Monocytes/Macrophages
4.8. Flow Cytometric Analysis
4.9. Cytospins
4.10. IL-6 Secretion Analysis
4.11. Induction of Apoptosis with the Chemical Inducer of Dimerization AP20187
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAVS1 | Adeno-associated virus integration site 1 |
| ADA | adenosine deaminase deficiency |
| alloHSCT | allogeneic hematopoietic stem cell transplantation |
| AMΦ | Alveolar macrophage |
| CAG | CMV early enhancer/chicken β-actin promoter |
| CID | chemical inducer of dimerization |
| DMEM | Dulbecco´s modified Eagle medium |
| EB | Embryoid body |
| ESC | Embryonic stem cells |
| herPAP | Hereditary pulmonary alveolar proteinosis |
| HSV-TK | Herpes Simplex virus thymidine kinase |
| iCasp9 | Inducible Caspase 9 |
| iPSC iMΦ | Induced pluripotent stem cells iPSC-derived macrophages |
| KO-DMEM | Knock Out Dulbecco´s modified Eagle medium |
| MCFC | Myeloid-cell forming complexes |
| MEF | murine embryonic fibroblast |
| MΦ | Macrophage |
| PB- MΦ | Peripheral blood-derived MΦ |
| PI | Propidium iodide |
| PMT | Pulmonary macrophage transplantation |
| TALEN | Transcription activator-like effector-based nucleases |
| TRM | Tissue-resident macrophage |
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Suga, M.; Kondo, T.; Inoue, H. Modeling Neurological Disorders with Human Pluripotent Stem Cell-Derived Astrocytes. Int. J. Mol. Sci. 2019, 20, 3862. [Google Scholar] [CrossRef]
- Kakinuma, S.; Watanabe, M. Analysis of the mechanism underlying liver diseases using human induced pluripotent stem cells. Immunol. Med. 2019, 42, 71–78. [Google Scholar] [CrossRef]
- Jang, J.; Yoo, J.-E.; Lee, J.-A.; Lee, D.R.; Kim, J.Y.; Huh, Y.J.; Kim, D.-S.; Park, C.-Y.; Hwang, D.-Y.; Kim, H.-S.; et al. Disease-specific induced pluripotent stem cells: A platform for human disease modeling and drug discovery. Exp. Mol. Med. 2012, 44, 202–213. [Google Scholar] [CrossRef]
- Attwood, S.; Edel, M. iPS-Cell Technology and the Problem of Genetic Instability—Can It Ever Be Safe for Clinical Use? J. Clin. Med. 2019, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Shimizu, K.; Matsumoto, R.; Honda, H. Selective Elimination of Human Induced Pluripotent Stem Cells Using Medium with High Concentration of L-Alanine. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Miki, K.; Parr, C.J.C.; Hayashi, K.; Takei, I.; Li, J.; Iwasaki, M.; Nakagawa, M.; Yoshida, Y.; Saito, H. Efficient, Selective Removal of Human Pluripotent Stem Cells via Ecto-Alkaline Phosphatase-Mediated Aggregation of Synthetic Peptides. Cell Chem. Biol. 2017, 24, 685–694.e4. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef]
- Hashimoto, H.; Kitano, S.; Ueda, R.; Ito, A.; Tada, K.; Fuji, S.; Yamashita, T.; Tomura, D.; Nukaya, I.; Mineno, J.; et al. Infusion of donor lymphocytes expressing the herpes simplex virus thymidine kinase suicide gene for recurrent hematologic malignancies after allogeneic hematopoietic stem cell transplantation. Int. J. Hematol. 2015, 102, 101–110. [Google Scholar] [CrossRef]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Renatus, M.; Stennicke, H.R.; Scott, F.L.; Liddington, R.C.; Salvesen, G.S. Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. USA 2001, 98, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Brenner, M.K. Improving the safety of T-Cell therapies using an inducible caspase-9 gene. Exp. Hematol. 2016, 44, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Itakura, G.; Kawabata, S.; Ando, M.; Nishiyama, Y.; Sugai, K.; Ozaki, M.; Iida, T.; Ookubo, T.; Kojima, K.; Kashiwagi, R.; et al. Fail-Safe System against Potential Tumorigenicity after Transplantation of iPSC Derivatives. Stem Cell Rep. 2017, 8, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Yagyu, S.; Hoyos, V.; Del Bufalo, F.; Brenner, M.K. An Inducible Caspase-9 Suicide Gene to Improve the Safety of Therapy Using Human Induced Pluripotent Stem Cells. Mol. Ther. 2015, 23, 1475–1485. [Google Scholar] [CrossRef]
- Ando, M.; Nishimura, T.; Yamazaki, S.; Yamaguchi, T.; Kawana-Tachikawa, A.; Hayama, T.; Nakauchi, Y.; Ando, J.; Ota, Y.; Takahashi, S.; et al. A safeguard system for induced pluripotent stem cell-derived rejuvenated T cell therapy. Stem Cell Rep. 2015, 5, 597–608. [Google Scholar] [CrossRef]
- Wu, C.; Hong, S.G.; Winkler, T.; Spencer, D.M.; Jares, A.; Ichwan, B.; Nicolae, A.; Guo, V.; Larochelle, A.; Dunbar, C.E. Development of an inducible caspase-9 safety switch for pluripotent stem cell–based therapies. Mol. Ther. Methods Clin. Dev. 2014, 1, 14053. [Google Scholar] [CrossRef]
- Yagyu, S.; Hoyos, V.; Del Bufalo, F.; Brenner, M.K. Multiple mechanisms determine the sensitivity of human-induced pluripotent stem cells to the inducible caspase-9 safety switch. Mol. Ther. Methods Clin. Dev. 2016, 3, 16003. [Google Scholar] [CrossRef]
- Ishida, M.; Miyagawa, S.; Saito, A.; Fukushima, S.; Harada, A.; Ito, E.; Ohashi, F.; Watabe, T.; Hatazawa, J.; Matsuura, K.; et al. Transplantation of Human-induced Pluripotent Stem Cell-derived Cardiomyocytes Is Superior to Somatic Stem Cell Therapy for Restoring Cardiac Function and Oxygen Consumption in a Porcine Model of Myocardial Infarction. Transplantation 2019, 103, 291–298. [Google Scholar] [CrossRef]
- Martin, U. Therapeutic Application of Pluripotent Stem Cells: Challenges and Risks. Front. Med. 2017, 4, 229. [Google Scholar] [CrossRef]
- Stoddard-Bennett, T.; Reijo Pera, R. Treatment of Parkinson’s Disease through Personalized Medicine and Induced Pluripotent Stem Cells. Cells 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, A.; Schultze, J.L. Tissue-resident macrophages—How to humanize our knowledge. Immunol. Cell Biol. 2017, 95, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Happle, C.; Lachmann, N.; Skuljec, J.; Wetzke, M.; Ackermann, M.; Brennig, S.; Mucci, A.; Jirmo, A.C.; Groos, S.; Mirenska, A.; et al. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci. Transl. Med. 2014, 6, 250ra113. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Arumugam, P.; Sakagami, T.; Lachmann, N.; Chalk, C.; Sallese, A.; Abe, S.; Trapnell, C.; Carey, B.; Moritz, T.; et al. Pulmonary macrophage transplantation therapy. Nature 2014, 514, 450–454. [Google Scholar] [CrossRef]
- Happle, C.; Lachmann, N.; Ackermann, M.; Mirenska, A.; Göhring, G.; Thomay, K.; Mucci, A.; Hetzel, M.; Glomb, T.; Suzuki, T.; et al. Pulmonary transplantation of human induced pluripotent stem cell-derived macrophages ameliorates pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2018, 198, 350–360. [Google Scholar] [CrossRef]
- Litvack, M.L.; Wigle, T.J.; Lee, J.; Wang, J.; Ackerley, C.; Grunebaum, E.; Post, M. Alveolar-like stem cell-derived myb2 macrophages promote recovery and survival in airway disease. Am. J. Respir. Crit. Care Med. 2016, 193, 1219–1229. [Google Scholar] [CrossRef]
- Haideri, S.S.; McKinnon, A.C.; Taylor, A.H.; Kirkwood, P.; Starkey Lewis, P.J.; O’Duibhir, E.; Vernay, B.; Forbes, S.; Forrester, L.M. Injection of embryonic stem cell derived macrophages ameliorates fibrosis in a murine model of liver injury. Npj. Regen. Med. 2017, 2, 1–10. [Google Scholar]
- Koba, C.; Haruta, M.; Matsunaga, Y.; Matsumura, K.; Haga, E.; Sasaki, Y.; Ikeda, T.; Takamatsu, K.; Nishimura, Y.; Senju, S. Therapeutic Effect of Human iPS-Cell-Derived Myeloid Cells Expressing IFN-β against Peritoneally Disseminated Cancer in Xenograft Models. PLoS ONE 2013, 8, e67567. [Google Scholar] [CrossRef]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kühnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Lachmann, N.; Ackermann, M.; Frenzel, E.; Liebhaber, S.; Brennig, S.; Happle, C.; Hoffmann, D.; Klimenkova, O.; Lüttge, D.; Buchegger, T.; et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Rep. 2015, 4, 282–296. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2012, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; Dekelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Cyranoski, D. “Reprogrammed” stem cells approved to mend human hearts for the first time news /631/532. Nature 2018, 557, 619–620. [Google Scholar] [CrossRef]
- Ogata, T.; Kozuka, T.; Kanda, T. Identification of an Insulator in AAVS1, a Preferred Region for Integration of Adeno-Associated Virus DNA. J. Virol. 2003, 77, 9000–9007. [Google Scholar] [CrossRef]
- Dreyer, A.K.; Hoffmann, D.; Lachmann, N.; Ackermann, M.; Steinemann, D.; Timm, B.; Siler, U.; Reichenbach, J.; Grez, M.; Moritz, T.; et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials 2015, 69, 191–200. [Google Scholar] [CrossRef]
- Kuhn, A.; Ackermann, M.; Mussolino, C.; Cathomen, T.; Lachmann, N.; Moritz, T. TALEN-mediated functional correction of human iPSC-derived macrophages in context of hereditary pulmonary alveolar proteinosis. Sci. Rep. 2017, 7, 15195. [Google Scholar] [CrossRef]
- Liang, Q.; Monetti, C.; Shutova, M.V.; Neely, E.J.; Hacibekiroglu, S.; Yang, H.; Kim, C.; Zhang, P.; Li, C.; Nagy, K.; et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature 2018, 563, 701–704. [Google Scholar] [CrossRef]
- Chen, F.; Cai, B.; Gao, Y.; Yuan, X.; Cheng, F.; Wang, T.; Jiang, M.; Zhou, Y.; Lahn, B.T.; Li, W.; et al. Suicide gene-mediated ablation of tumor-initiating mouse pluripotent stem cells. Biomaterials 2013, 34, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, M.; Pastorino, F.; Di Paolo, D.; Perri, P.; Brignole, C. Targeting macrophages as a potential therapeutic intervention: Impact on inflammatory diseases and cancer. Int. J. Mol. Sci. 2018, 19, 1953. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kivimäe, S.; Dolor, A.; Szoka, F.C. Macrophage-based cell therapies: The long and winding road. J. Control. Release 2016, 240, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Rodermond, H.; Colak, S.; Grandela, C.; Medema, J.P. Targeting colorectal cancer stem cells with inducible caspase-9. Apoptosis 2012, 17, 528–537. [Google Scholar] [CrossRef]
- Iuliucci, J.D.; Oliver, S.D.; Morley, S.; Ward, C.; Ward, J.; Dalgarno, D.; Clackson, T.; Berger, H.J. Intravenous safety and pharmacokinetics of a novel dimerizer drug, AP1903, in healthy volunteers. J. Clin. Pharmacol. 2001, 41, 870–879. [Google Scholar] [CrossRef]
- Lachmann, N.; Happle, C.; Ackermann, M.; L??ttge, D.; Wetzke, M.; Merkert, S.; Hetzel, M.; Kensah, G.; Jara-Avaca, M.; Mucci, A.; et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014, 189, 167–182. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipus, A.; Janosz, E.; Ackermann, M.; Hetzel, M.; Dahlke, J.; Buchegger, T.; Wunderlich, S.; Martin, U.; Cathomen, T.; Schambach, A.; et al. Targeted Integration of Inducible Caspase-9 in Human iPSCs Allows Efficient in vitro Clearance of iPSCs and iPSC-Macrophages. Int. J. Mol. Sci. 2020, 21, 2481. https://doi.org/10.3390/ijms21072481
Lipus A, Janosz E, Ackermann M, Hetzel M, Dahlke J, Buchegger T, Wunderlich S, Martin U, Cathomen T, Schambach A, et al. Targeted Integration of Inducible Caspase-9 in Human iPSCs Allows Efficient in vitro Clearance of iPSCs and iPSC-Macrophages. International Journal of Molecular Sciences. 2020; 21(7):2481. https://doi.org/10.3390/ijms21072481
Chicago/Turabian StyleLipus, Alexandra, Ewa Janosz, Mania Ackermann, Miriam Hetzel, Julia Dahlke, Theresa Buchegger, Stephanie Wunderlich, Ulrich Martin, Toni Cathomen, Axel Schambach, and et al. 2020. "Targeted Integration of Inducible Caspase-9 in Human iPSCs Allows Efficient in vitro Clearance of iPSCs and iPSC-Macrophages" International Journal of Molecular Sciences 21, no. 7: 2481. https://doi.org/10.3390/ijms21072481
APA StyleLipus, A., Janosz, E., Ackermann, M., Hetzel, M., Dahlke, J., Buchegger, T., Wunderlich, S., Martin, U., Cathomen, T., Schambach, A., Moritz, T., & Lachmann, N. (2020). Targeted Integration of Inducible Caspase-9 in Human iPSCs Allows Efficient in vitro Clearance of iPSCs and iPSC-Macrophages. International Journal of Molecular Sciences, 21(7), 2481. https://doi.org/10.3390/ijms21072481