Generation of An Endogenous FGFR2–BICC1 Gene Fusion/58 Megabase Inversion Using Single-Plasmid CRISPR/Cas9 Editing in Biliary Cells

 and
and
Abstract
1. Introduction
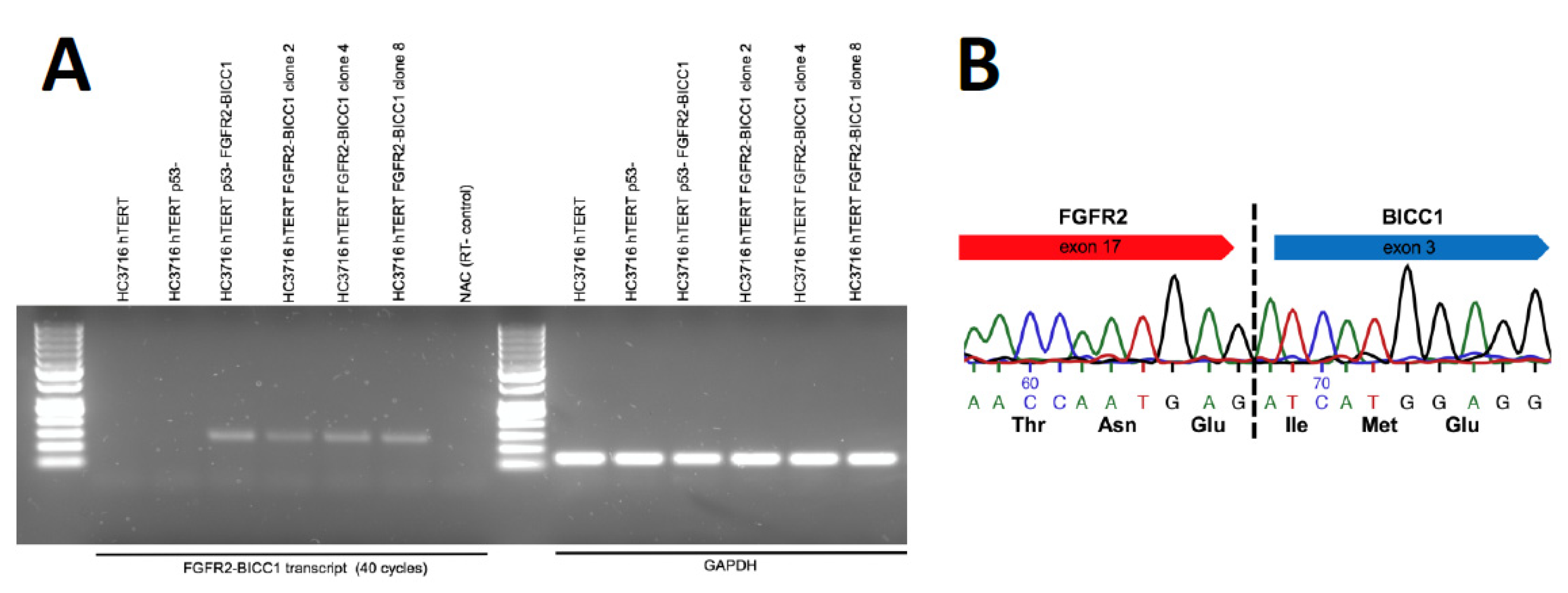
2. Results
3. Discussion
4. Materials and Methods
4.1. Construction of CRISPR Plasmids
4.2. Transfection
4.3. Isolation of Single Cells
4.4. Detection of Genomic Rearrangement by PCR
4.5. T7 Endonuclease Assay
4.6. RNA Isolation and RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Borad, M.J.; Gores, G.J.; Roberts, L.R. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr. Opin. Gastroenterol. 2015, 31, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Peiris, M.N.; Donoghue, D.J. Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma. Cytokine Growth Factor Rev. 2019, 52, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.J.; Zhang, Y.; Ercan, R.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bempt, M.V.; Demeyer, S.; Mentens, N.; Geerdens, E.; De Bock, C.E.; Wlodarska, I.; Cools, J. Generation of the Fip1l1–Pdgfra fusion gene using CRISPR/Cas genome editing. Nat. Publ. Group 2016, 30, 1913–1916. [Google Scholar]
- Blasco, R.B.; Karaca, E.; Ambrogio, C.; Cheong, T.C.; Karayol, E.; Minero, V.G.; Chiarle, R. Simple and Rapid In Vivo Generation of Chromosomal Rearrangements using CRISPR/Cas9 Technology. CellReports 2014, 9, 1219–1227. [Google Scholar]
- Choi, P.S.; Meyerson, M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Reimer, J.; Knöß, S.; Labuhn, M.; Charpentier, E.M.; Göhring, G.; Schlegelberger, B.; Klusmann, J.H.; Heckl, D. CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate leukemia in human hematopoietic progenitor cells in vivo. Haematologica 2017, 102, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.; Martin, M.C.; Garcia, A.; Cigudosa, J.C.; Ramirez, J.C.; Rodriguez-Perales, S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014, 5, 3964. [Google Scholar] [CrossRef] [PubMed]
- Waki, K.; Anno, K.; Ono, T.; Ide, T.; Chayama, K.; Tahara, H. Establishment of functional telomerase immortalized human hepatocytes and a hepatic stellate cell line for telomere-targeting anticancer drug development. Cancer Sci. 2010, 101, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Kwong, L.N. Insights into the origin of intrahepatic cholangiocarcinoma from mouse models. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Van Chu, T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef] [PubMed]
- Essletzbichler, P.; Konopka, T.; Santoro, F.; Chen, D.; Gapp, B.V.; Kralovics, R.; Brummelkamp, T.R.; Nijman, S.M.B.; Burckstummer, T. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014, 24, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Plasmid System | # Colonies Screened | # Colonies Positive | % Colonies Positive | # +Colonies Expressing Fusion |
|---|---|---|---|---|---|
| Hc3716 | 2 plasmids | 47 | 2 | 4.2% | 2/2 |
| Hc3716 | 1 plasmid | 66 | 2 | 3.0% | 2/2 |
| Hc3716 shp53 | 1 plasmid | 70 | 1 | 1.4% | 1/1 |
| HUH-28 | 1 plasmid | 32 | 5 | 15% | 1/3 |
| MMNK-1 | 1 plasmid | 32 | 2 | 6.2% | 2/2 |
| CCSW-1 | 1 plasmid | 155 | 15 | 9.7% | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reicher, A.; Harris, A.L.; Prinz, F.; Kiesslich, T.; Wei, M.; Öllinger, R.; Rad, R.; Pichler, M.; Kwong, L.N. Generation of An Endogenous FGFR2–BICC1 Gene Fusion/58 Megabase Inversion Using Single-Plasmid CRISPR/Cas9 Editing in Biliary Cells. Int. J. Mol. Sci. 2020, 21, 2460. https://doi.org/10.3390/ijms21072460
Reicher A, Harris AL, Prinz F, Kiesslich T, Wei M, Öllinger R, Rad R, Pichler M, Kwong LN. Generation of An Endogenous FGFR2–BICC1 Gene Fusion/58 Megabase Inversion Using Single-Plasmid CRISPR/Cas9 Editing in Biliary Cells. International Journal of Molecular Sciences. 2020; 21(7):2460. https://doi.org/10.3390/ijms21072460
Chicago/Turabian StyleReicher, Andreas, Antoneicka L Harris, Felix Prinz, Tobias Kiesslich, Miaoyan Wei, Rupert Öllinger, Roland Rad, Martin Pichler, and Lawrence N Kwong. 2020. "Generation of An Endogenous FGFR2–BICC1 Gene Fusion/58 Megabase Inversion Using Single-Plasmid CRISPR/Cas9 Editing in Biliary Cells" International Journal of Molecular Sciences 21, no. 7: 2460. https://doi.org/10.3390/ijms21072460
APA StyleReicher, A., Harris, A. L., Prinz, F., Kiesslich, T., Wei, M., Öllinger, R., Rad, R., Pichler, M., & Kwong, L. N. (2020). Generation of An Endogenous FGFR2–BICC1 Gene Fusion/58 Megabase Inversion Using Single-Plasmid CRISPR/Cas9 Editing in Biliary Cells. International Journal of Molecular Sciences, 21(7), 2460. https://doi.org/10.3390/ijms21072460