Novel Autoantibody Signatures in Sera of Patients with Pancreatic Cancer, Chronic Pancreatitis and Autoimmune Pancreatitis: A Protein Microarray Profiling Approach

 , , , and
, , , and
Abstract
1. Introduction
2. Results
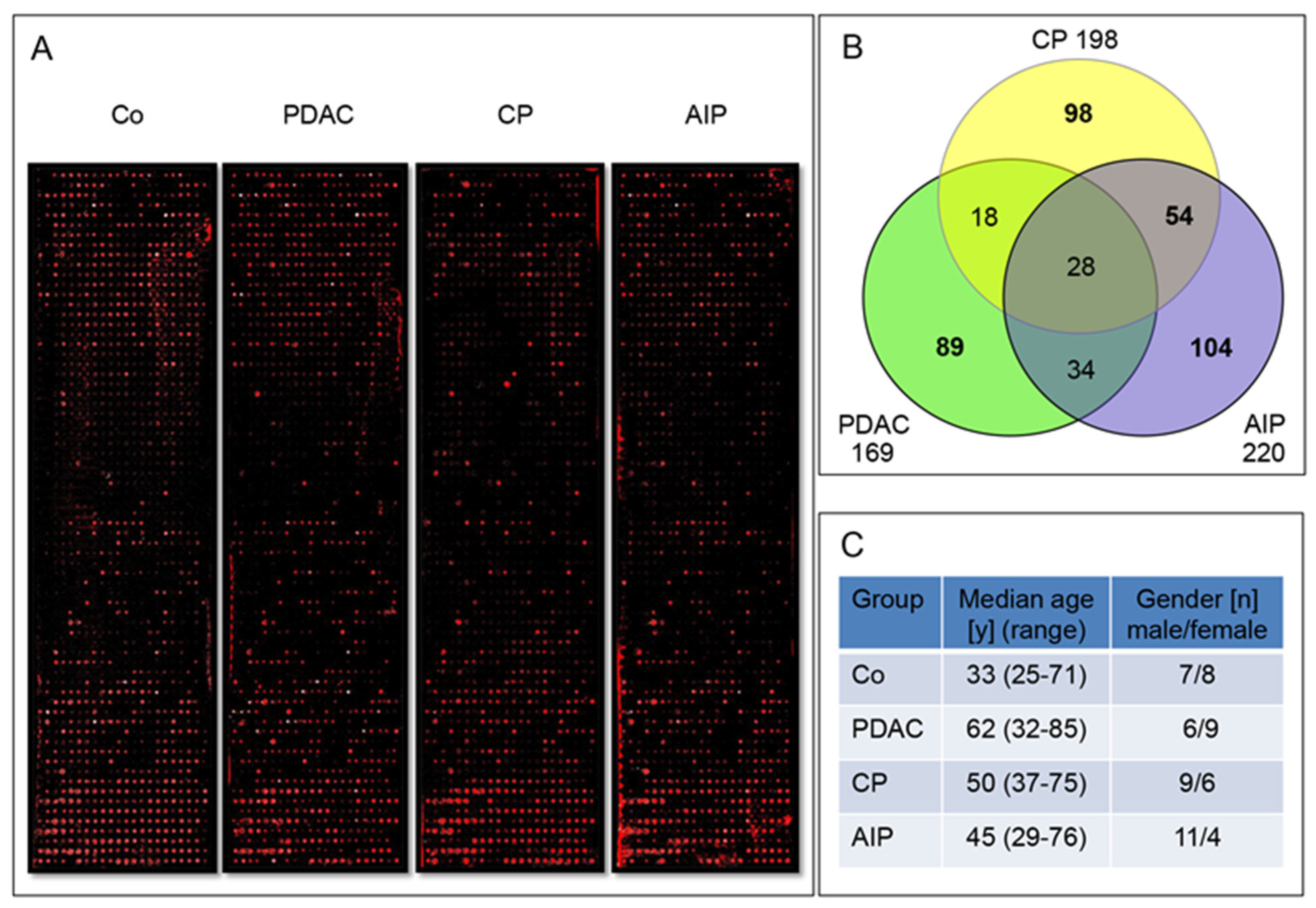
2.1. Screening of Pooled IgG Enriched Sera Fractions on Large Protein Microarrays
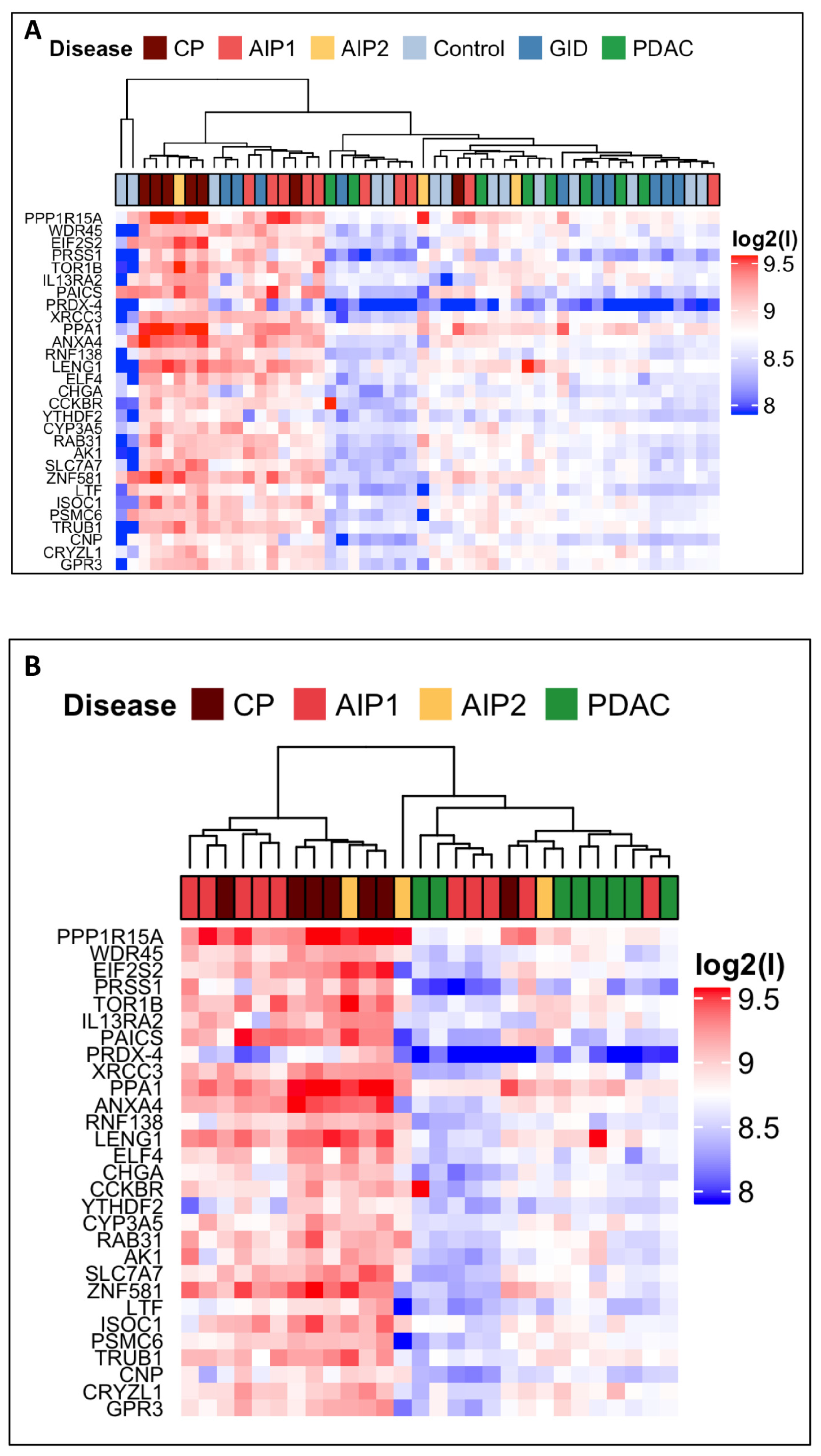
2.2. Screening of Multiple Sera of a Large Cohort
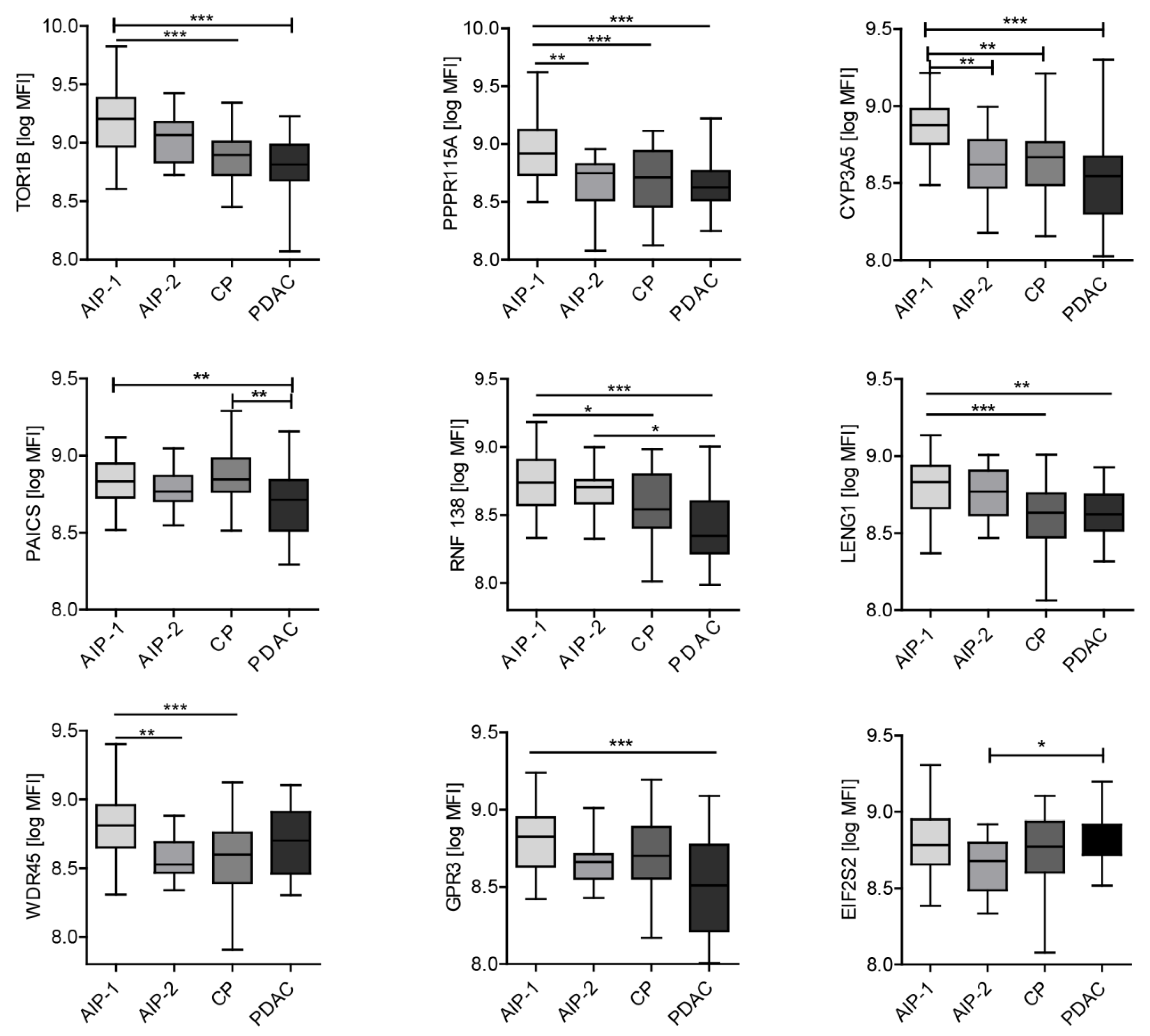
2.3. Autoantibody Profiling of Selected Autoantigens with Individual Samples
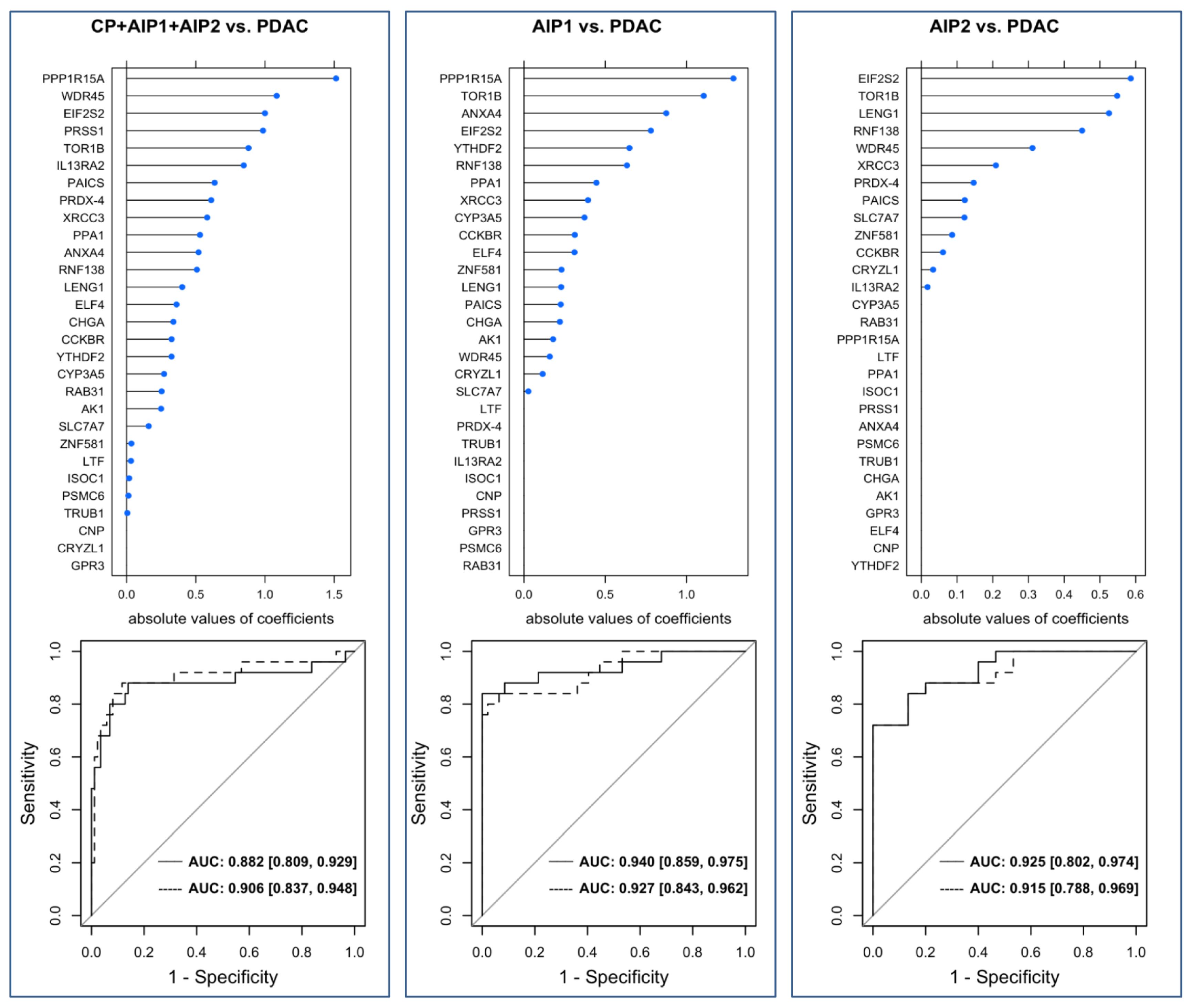
2.4. Validation of the Marker Panel
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. IgG Enrichment
4.3. Generation of DNA Templates for Recombinant Human Antigens from Bacterial Library
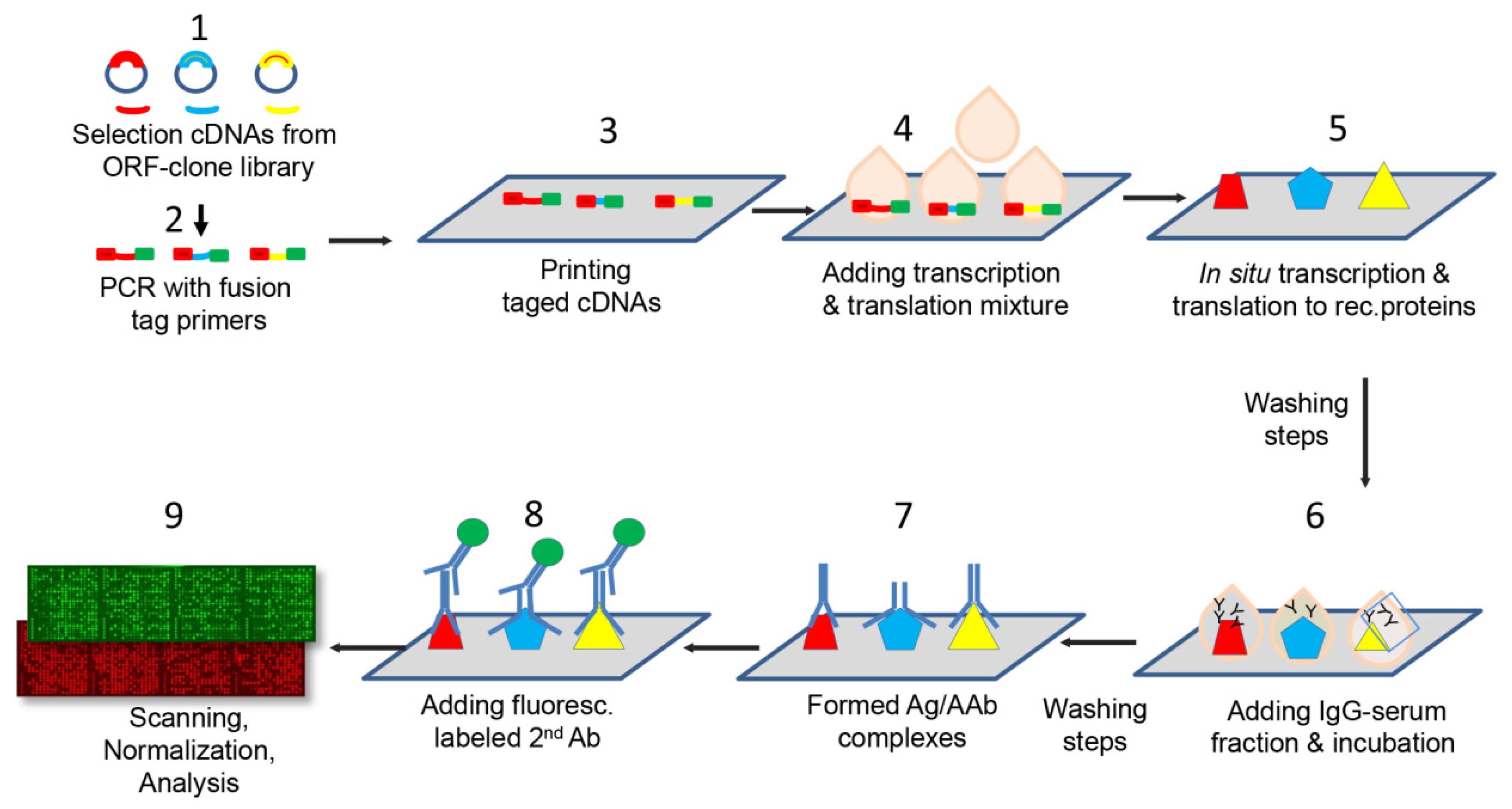
4.4. Manufacture of Protein Microarrays
4.5. Immunoassay with Patient Antibodies
4.6. Construction and Optimization of the Protein Microarrays
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAb | Autoantibody |
| AIP | Autoimmune pancreatitis |
| AUC | Area under the curve |
| CP | Chronic pancreatitis |
| GID | Gastrointestinal diseases |
| EBV VCA p18 Epstein | Barr Virus Capsid Antigen p 1 |
| MFI | Median fluorescence intensity |
| PDAC | Pancreatic ductal adenocarcinoma |
References
- De Castro, S.M.; De Nes, L.C.; Nio, C.Y.; Velseboer, D.C.; Ten Kate, F.J.; Busch, O.R.; Van Gulik, T.M.; Gouma, D.J. Incidence and characteristics of chronic and lymphoplasmacytic sclerosing pancreatitis in patients scheduled to undergo a pancreatoduodenectomy. HPB 2010, 12, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hardacre, J.M.; Iacobuzio-Donahue, C.A.; Sohn, T.A.; Abraham, S.C.; Yeo, C.J.; Lillemoe, K.D.; Choti, M.A.; Campbell, K.A.; Schulick, R.D.; Hruban, R.H.; et al. Results of pancreaticoduodenectomy for lymphoplasmacytic sclerosing pancreatitis. Ann. Surg. 2003, 237, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.M.; Cubukcu-Dimopulo, O.; Palesty, J.A.; Suriawinata, A.; Klimstra, D.; Brennan, M.F.; Conlon, K. Lymphoplasmacytic sclerosing pancreatitis: Inflammatory mimic of pancreatic carcinoma. J. Gastrointest. Surg. 2003, 7, 129–137. [Google Scholar] [CrossRef]
- Kamisawa, T.; Chari, S.T.; Giday, S.A.; Kim, M.H.; Chung, J.B.; Lee, K.T.; Werner, J.; Bergmann, F.; Lerch, M.M.; Mayerle, J.; et al. Clinical profile of autoimmune pancreatitis and its histological subtypes: An international multicenter survey. Pancreas 2011, 40, 809–814. [Google Scholar] [CrossRef]
- Ruckert, F.; Pilarsky, C.; Grutzmann, R. Serum tumor markers in pancreatic cancer-recent discoveries. Cancers 2010, 2, 1107–1124. [Google Scholar] [CrossRef]
- Hamanaka, Y.; Hamanaka, S.; Suzuki, M. Sialyl Lewis(a) ganglioside in pancreatic cancer tissue correlates with the serum CA 19-9 level. Pancreas 1996, 13, 160–165. [Google Scholar] [CrossRef]
- Felix, K.; Hauck, O.; Schnölzer, M.; Kempf, T.; Warnken, U.; Schneider, K.; Bergmann, F.; Fritz, S.; Werner, J. Identification of Novel Serum Autoantibodies for Differential Diagnosis of Autoimmune Pancreatitis and Pancreatic Ductal Adenocarcinoma. Pancreas 2016, 45, 1309–1319. [Google Scholar] [CrossRef]
- Gnjatic, S.; Ritter, E.; Büchler, M.W.; Giese, N.A.; Brors, B.; Frei, C.; Murray, A.; Halama, N.; Zörnig, I.; Chen, Y.T.; et al. Seromic profiling of ovarian and pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 5088–5093. [Google Scholar] [CrossRef]
- Tomaino, B.; Cappello, P.; Capello, M.; Fredolini, C.; Ponzetto, A.; Novarino, A.; Ciuffreda, L.; Bertetto, O.; De Angelis, C.; Gaia, E.; et al. Autoantibody signature in human ductal pancreatic adenocarcinoma. J. Proteome Res. 2007, 6, 4025–4031. [Google Scholar] [CrossRef]
- Dumstrei, K.; Chen, H.; Brenner, H. A systematic review of serum autoantibodies as biomarkers for pancreatic cancer detection. Oncotarget 2016, 7, 11151–11164. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Song, W.; Fang, Y.; Yu, L.; Liu, S.; Churilov, L.P.; Zhang, F. The roles and applications of autoantibodies in progression, diagnosis, treatment and prognosis of human malignant tumours. Autoimmun. Rev. 2017, 16, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Gure, A.O.; Scanlan, M.J. Serological analysis of expression cDNA libraries (SEREX): An immunoscreening technique for identifying immunogenic tumor antigens. Methods Mol. Med. 2005, 103, 207–216. [Google Scholar] [PubMed]
- Jaras, K.; Anderson, K. Autoantibodies in cancer: Prognostic biomarkers and immune activation. Expert Rev. Proteom. 2011, 8, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef]
- Zaenker, P.; Gray, E.S.; Ziman, M.R. Autoantibody Production in Cancer--The Humoral Immune Response toward Autologous Antigens in Cancer Patients. Autoimmun. Rev. 2016, 15, 477–483. [Google Scholar] [CrossRef]
- Betzen, C.; Alhamdani, M.S.; Lueong, S.; Schroder, C.; Stang, A.; Hoheisel, J.D. Clinical proteomics: Promises, challenges and limitations of affinity arrays. Proteom. Clin. Appl. 2015, 9, 342–347. [Google Scholar] [CrossRef]
- Angenendt, P.; Kreutzberger, J.; Glokler, J.; Hoheisel, J.D. Generation of high density protein microarrays by cell-free in situ expression of unpurified PCR products. Mol. Cell Proteom. 2006, 5, 1658–1666. [Google Scholar] [CrossRef]
- Smyk, D.S.; Rigopoulou, E.I.; Koutsoumpas, A.L.; Kriese, S.; Burroughs, A.K.; Bogdanos, D.P. Autoantibodies in autoimmune pancreatitis. Int. J. Rheumatol. 2012, 2012, 940831. [Google Scholar] [CrossRef]
- Wilson, E.B. Probable Inference, the Law of Succession, and Statistical Inference. J. Am. Stat. Assoc. 1027, 22, 209–212. [Google Scholar] [CrossRef]
- Canzler, U.; Bartsch, H.; Ulitzsch, S.; Kurien, B.T.; Dorri, Y.; Scofield, R.H.; Grossmann, K.; Lehmann, W.; Pilarsky, C.; Denz, A.; et al. Detection of autoantibodies to tumour-associated antigens in sera of patients with systemic autoimmunity using a novel protein microblot array. Scand. J. Immunol. 2009, 69, 563–569. [Google Scholar] [CrossRef]
- Kijanka, G.; Murphy, D. Protein arrays as tools for serum autoantibody marker discovery in cancer. J. Proteom. 2009, 72, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Poetz, O.; Schwenk, J.M.; Kramer, S.; Stoll, D.; Templin, M.F.; Joos, T.O. Protein microarrays: Catching the proteome. Mech. Ageing Dev. 2005, 126, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Végh, P.; Miklós, K.; Németh, J.; Rásky, K.; Péterfy, F.; Erdei, A.; Prechl, J. Detection of complement activation on antigen microarrays generates functional antibody profiles and helps characterization of disease-associated changes of the antibody repertoire. J. Immunol. 2008, 181, 8162–8169. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Szekeres, Z.; Erdei, A.; Prechl, J. Two-dimensional immune profiles improve antigen microarray-based characterization of humoral immunity. Proteomics 2008, 8, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Babel, I.; Barderas, R.; Diaz-Uriarte, R.; Martinez-Torrecuadrada, J.L.; Sanchez-Carbayo, M.; Casal, J.I. Identification of tumor-associated autoantigens for the diagnosis of colorectal cancer in serum using high density protein microarrays. Mol. Cell Proteom. 2009, 8, 2382–2395. [Google Scholar] [CrossRef]
- Orenes-Piñero, E.; Barderas, R.; Rico, D.; Casal, J.I.; Gonzalez-Pisano, D.; Navajo, J.; Algaba, F.; Piulats, J.M.; Sanchez-Carbayo, M. Serum and tissue profiling in bladder cancer combining protein and tissue arrays. J. Proteome Res. 2010, 9, 164–173. [Google Scholar] [CrossRef]
- Hudson, M.E.; Pozdnyakova, I.; Haines, K.; Mor, G.; Snyder, M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc. Natl. Acad. Sci. USA 2007, 104, 17494–17499. [Google Scholar] [CrossRef]
- Okazaki, K.; Uchida, K.; Ohana, M.; Nakase, H.; Uose, S.; Inai, M.; Matsushima, Y.; Katamura, K.; Ohmori, K.; Chiba, T. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000, 118, 573–581. [Google Scholar] [CrossRef]
- Uchida, K.; Okazaki, K.; Konishi, Y.; Ohana, M.; Takakuwa, H.; Hajiro, K.; Chiba, T. Clinical analysis of autoimmune-related pancreatitis. Am. J. Gastroenterol. 2000, 95, 2788–2794. [Google Scholar] [CrossRef]
- Löhr, J.M.; Faissner, R.; Koczan, D.; Bewerunge, P.; Bassi, C.; Brors, B.; Eils, R.; Frulloni, L.; Funk, A.; Halangk, W.; et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: Gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am. J. Gastroenterol. 2010, 105, 2060–2071. [Google Scholar] [CrossRef]
- Deshpande, V.; Gupta, R.; Sainani, N.; Sahani, D.V.; Virk, R.; Ferrone, C.; Khosroshahi, A.; Stone, J.H.; Lauwers, G.Y. Subclassification of autoimmune pancreatitis: A histologic classification with clinical significance. Am. J. Surg. Pathol. 2011, 35, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kanno, A.; Masamune, A.; Okazaki, K.; Kamisawa, T.; Kawa, S.; Nishimori, I.; Tsuji, I.; Shimosegawa, T. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2011. Pancreas 2015, 44, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.P.; Chari, S.T.; Pannala, R.; Sugumar, A.; Clain, J.E.; Levy, M.J.; Pearson, R.K.; Smyrk, T.C.; Petersen, B.T.; Topazian, M.D.; et al. Differences in clinical profile and relapse rate of type 1 versus type 2 autoimmune pancreatitis. Gastroenterology 2010, 139, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Okazaki, K.; Nishi, T.; Uose, S.; Nakase, H.; Ohana, M.; Matsushima, Y.; Omori, K.; Chiba, T. Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Lab Invest. 2002, 82, 411–424. [Google Scholar] [CrossRef]
- Chen, R.; Brentnall, T.A.; Pan, S.; Cooke, K.; Moyes, K.W.; Lane, Z.; Crispin, D.A.; Goodlett, D.R.; Aebersold, R.; Bronner, M.P. Quantitative proteomics analysis reveals that proteins differentially expressed in chronic pancreatitis are also frequently involved in pancreatic cancer. Mol. Cell Proteom. 2007, 6, 1331–1342. [Google Scholar] [CrossRef]
- Wiemann, S.; Pennacchio, C.; Hu, Y.; Hunter, P.; Harbers, M.; Amiet, A.; Bethel, G.; Busse, M.; Carninci, P.; Dunham, I.; et al. The ORFeome Collaboration: A genome-scale human ORF-clone resource. Nat. Methods 2016, 13, 191–192. [Google Scholar]
- Syafrizayanti Lueong, S.S.; Di, C.; Schaefer, J.V.; Pluckthun, A.; Hoheisel, J.D. Personalised proteome analysis by means of protein microarrays made from individual patient samples. Sci. Rep. 2017, 7, 39756. [Google Scholar] [CrossRef]
- Hufnagel, K.; Lueong, S.; Willhauck-Fleckenstein, M.; Hotz-Wagenblatt, A.; Miao, B.; Bauer, A.; Michel, A.; Butt, J.; Pawlita, M.; Hoheisel, J.D.; et al. Immunoprofiling of Chlamydia trachomatis using whole-proteome microarrays generated by on-chip in situ expression. Sci. Rep. 2018, 8, 7503. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Kuhn, M.; Jed Wing, S.W.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; Benesty, M.; Lescarbeau, R.; et al. Caret: Classification and Regression Training. R Package Version 6.0-84. 2019. Available online: https://CRANR-projectorg/package=caret (accessed on 19 December 2019).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Xu, W.; Ji, S.; Zhang, B.; Ni, Q.; Xu, J.; Yu, X. Diagnostic and prognostic value of carcinoembryonic antigen in pancreatic cancer: A systematic review and meta-analysis. OncoTargets Ther. 2017, 10, 4591–4598. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients Type | Sample Set | Patients (n) | Median Age (Range) | Male (n) (%) |
|---|---|---|---|---|
| Co | 1 | 15 | 33.0 (25–71) | 7 (46.7) |
| 2 | 70 | 40.5 (20–83) | 29 (70.0) | |
| 3 | 48 | 48.5 (20–81) | 31 (64.6) | |
| PDAC | 1 | 15 | 66.0 (54–77) | 6 (40.0) |
| 2 | 40 | 68.0 (32–85) | 22 (55.0) | |
| 3 | 25 | 68.0 (41–85) | 15 (60.0) | |
| CP | 1 | 15 | 55.0 (37–75) | 9 (60.0) |
| 2 | 35 | 52.0 (36–68) | 21 (60.0) | |
| 3 | 24 | 53.5 (36–68) | 16 (66.7) | |
| GID | 1 | 0 | 0 | 0 |
| 2 | 50 | 63.5 (25–83) | 33 (66.0) | |
| 3 | 26 | 63.0 (43–81)) | 19 (73.1) | |
| AIP-1 | 1 | 8 | 56.5 (29–76) | 6 (75.0) |
| 2 | 50 | 68.0 (29–84) | 38 (76.0) | |
| 3 | 47 | 65.0 (29–83) | 35 (74.5) | |
| AIP-2 | 1 | 7 | 43.0 (37–67) | 5 (71.4) |
| 2 | 15 | 45.0 (32–76) | 11 (73.3) | |
| 3 | 15 | 45.0 (32–76) | 11 (73.3) | |
| All patients | 1 | 60 | 63.0 (25–77) | 33 (55.0) |
| 2 | 260 | 58.0 (20–85) | 154 (59.2) | |
| 3 | 185 | 59.0 (24–85) | 127 (68.6) |
| Gene Symbol | Antigen Description | ORF Length (bp) |
|---|---|---|
| CCKBR | Cholecystokinin B receptor | 1341 |
| CRYZL1 | Crystallin, zeta (quinone reductase)-like 1 | 1050 |
| TRUB1 | TruB pseudouridine (psi) synthase homolog 1 (Escherichia coli) | 1047 |
| WDR45 | WD repeat domain 45 | 1083 |
| CYP3A5 | Cytochrome P450, family 3, subfamily A, polypeptide 5 | 1509 |
| IL13RA2 | Interleukin 13 receptor, alpha 2 | 1140 |
| ANXA4 | Annexin A4 | 963 |
| PAICS | Phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoimidazole succino-carboxamide synthase | 1278 |
| EIF2S2 | Eukaryotic translation initiation factor 2, subunit 2 beta, 38 kDa | 1002 |
| SLC7A7 | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 7 | 1536 |
| RNF138 | Ring finger protein 138 | 738 |
| CNP | 2’,3’-cyclic nucleotide 3’ phosphodiesterase | 1263 |
| AK1 | Adenylate kinase 1 | 585 |
| YTHDF2 | YTH domain family, member 2 | 1740 |
| ELF4 | E74-like factor 4 (ets domain transcription factor) | 1992 |
| RAB31 | RAB31, member RAS oncogene family | 585 |
| CHGA | Chromogranin A (parathyroid secretory protein 1) | 1374 |
| PSMC6 | Proteasome (prosome, macropain) 26S subunit, ATPase, 6 | 1167 |
| GPR3 | G protein-coupled receptor 3 | 993 |
| TOR1B | Torsin family 1, member B (torsin B) | 1011 |
| XRCC3 | X-ray repair complementing defective repair in Chinese hamster cells 3 | 1038 |
| ISOC1 | Isochorismatase domain containing 1 | 708 |
| LENG1 | Leukocyte receptor cluster (LRC) member 1 | 792 |
| PPA1 | Pyrophosphatase (inorganic) 1 | 870 |
| ZNF581 | Zinc finger protein 581 | 594 |
| PRSS1 | Protease, serine, 1 (trypsin 1) | 720 |
| PPP1R15A | Protein phosphatase 1, regulatory (inhibitor) subunit 15A | 2025 |
| LTF | Lactotransferrin | 2136 |
| PRDX-4 | Peroxiredoxin 4 | 813 |
| Gene Symbol | Co | PDAC | CP | GID | AIP-1 | AIP-2 |
|---|---|---|---|---|---|---|
| CCKBR | 8.6305 | 8.7447 | 9.0325 | 8.6781 | 8.7038 | 8.9954 |
| CRYZL1 | 8.7371 | 8.8043 | 8.9705 | 8.7381 | 8.8542 | 8.8528 |
| TRUB1 | 8.7193 | 8.8316 | 9.1490 | 8.7414 | 8.7844 | 8.9571 |
| WDR45 | 8.6397 | 8.7019 | 9.0841 | 8.8597 | 8.8556 | 8.8729 |
| CYP3A5 | 8.6718 | 8.7179 | 9.0051 | 8.7582 | 8.7711 | 8.8270 |
| IL13RA2 | 8.6985 | 8.7315 | 9.0586 | 8.7002 | 8.7162 | 8.9898 |
| ANXA4 | 8.6938 | 8.7661 | 9.3695 | 8.7482 | 8.9397 | 8.8128 |
| PAICS | 8.6322 | 8.5264 | 9.2560 | 8.5043 | 9.0537 | 9.0789 |
| EIF2S2 | 8.7413 | 8.6949 | 9.2072 | 8.6407 | 8.8949 | 8.7997 |
| SLC7A7 | 8.6244 | 8.6772 | 9.1777 | 8.6259 | 8.7772 | 8.8422 |
| RNF138 | 8.6440 | 8.6127 | 9.0573 | 8.5893 | 8.6385 | 9.0285 |
| CNP | 8.5802 | 8.5065 | 8.8401 | 8.4649 | 8.5370 | 8.9146 |
| AK1 | 8.6401 | 8.6145 | 8.9979 | 8.5715 | 8.5416 | 8.9238 |
| YTHDF2 | 8.4544 | 8.5643 | 8.8103 | 8.5429 | 8.4154 | 8.8491 |
| ELF4 | 8.6501 | 8.6388 | 9.0962 | 8.7032 | 8.6739 | 8.8977 |
| RAB31 | 8.6347 | 8.6322 | 9.0612 | 8.7034 | 8.7708 | 9.0932 |
| CHGA | 8.5721 | 8.5743 | 9.0147 | 8.5691 | 8.6180 | 8.6760 |
| PSMC6 | 8.6986 | 8.7140 | 9.0575 | 8.6634 | 8.7957 | 8.7146 |
| GPR3 | 8.7187 | 8.7172 | 9.0569 | 8.7065 | 8.7890 | 8.7467 |
| TOR1B | 8.6135 | 8.7880 | 9.1709 | 8.7872 | 8.8545 | 8.9956 |
| XRCC3 | 8.6368 | 8.7289 | 9.1951 | 8.7563 | 8.7511 | 9.1535 |
| ISOC1 | 8.6692 | 8.7422 | 9.0783 | 8.7196 | 8.7657 | 8.9872 |
| LENG1 | 8.8537 | 8.8802 | 9.3380 | 8.8457 | 8.8833 | 8.8929 |
| PPA1 | 8.9197 | 8.90522 | 9.5752 | 8.8729 | 9.1075 | 9.1996 |
| ZNF581 | 8.9016 | 8.6838 | 9.2369 | 8.6845 | 9.0998 | 8.9404 |
| PRSS1 | 8.4126 | 8.3032 | 8.9181 | 8.3791 | 8.4931 | 8.7592 |
| PPP1R15A | 8.8223 | 8.7876 | 9.5294 | 8.8394 | 9.1736 | 9.4495 |
| LTF | 8.5557 | 8.5745 | 8.906 | 8.4974 | 8.6029 | 8.4205 |
| PRDX-4 | 7.9297 | 8.0862 | 8.7052 | 8.1987 | 8.0611 | 8.3418 |
| Gene Symbol | PDAC | AIP | p Value |
|---|---|---|---|
| PAICS | 6091 | 6765 | 0.0436 |
| BOK | 6324 | 8439 | 0.0025 |
| RNF138 | 4214 | 6352 | 0.0016 |
| TOR1B | 6727 | 10,077 | 5.56 × 10−6 |
| PPP1R15A | 5576 | 6458 | 0.0017 |
| LENG1 | 5560 | 6391 | 0.0016 |
| CYP3A5 | 5141 | 6717 | 0.0075 |
| GPR3 | 4970 | 6481 | 0.0137 |
| CA2 | 5576 | 4667 | 0.0173 |
| HDAC3 | 4214 | 6352 | 0.0016 |
| Variables | Patients (n) |
|---|---|
| Total | 315 |
| Pancreatic cancer (PDAC) | 65 |
| Age (years) | |
| Mean ± SD | 66.2 ± 11 |
| Range (median) | 32–85 (68) |
| Gender: male/female | 32/33 |
| Grade | |
| G1 | 0 |
| - G2 | 32 |
| - G3 | 33 |
| TNM-stage (AJCC stage, 8th edition) | |
| - T1a | 1 |
| - T1c | 10 |
| - T2 | 34 |
| - T3 | 14 |
| - T3 * | 6 |
| N-stage | |
| - N0 | 11 |
| - N1 | 26 |
| - N1* | 3 |
| - N2 | 25 |
| M-stage | |
| - M0 | 53 |
| - M1 | 12 |
| AJCC Stage | |
| - IA | 5 |
| - IB | 6 |
| - IIA | 2 |
| - IIB | 19 |
| - IIB * | 3 |
| - III | 18 |
| - IV | 12 |
| Tumour location | |
| - head | 42 |
| - body | 8 |
| - tail | 15 |
| Autoimmune pancreatitis (AIP) | 70 |
| AIP type 1 | 55 |
| Age (years) | |
| Mean ±SD | 64.47 ± 12.21 |
| Range (median) | 29–84 (68) |
| Gender: male/female | 42/13 |
| AIP type 2 | 15 |
| Age (years) | |
| Mean ±SD | 51.86 ± 14.7 |
| Range (median) | 32–76 (44.5) |
| Gender: male/female | 10/5 |
| Chronic pancreatitis (CP) | 50 |
| Age (years) | |
| Mean ±SD | 53.34 ± 9.6 |
| Range (median) | 36–75 (53.5) |
| Gender: male/female | 33/17 |
| Gastrointestinal diseases (GID) | 60 |
| Benign diseases | 11 |
| Malign diseases | 49 |
| - gastro, liver, colon, renal, other | 6, 6, 17, 4, 16 |
| Age (years) | |
| Mean ±SD | 63.28 ± 10.75 |
| Range (median) | 25–83 (63) |
| Gender: male/female | 41/19 |
| Healthy controls (Co) | 70 |
| Age (years) | |
| Mean ±SD | 46.8 ± 19.47 |
| Range (median) | 20–83 (40.5) |
| Gender: male/female | 48/22 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghassem-Zadeh, S.; Hufnagel, K.; Bauer, A.; Frossard, J.-L.; Yoshida, M.; Kutsumi, H.; Acha-Orbea, H.; Neulinger-Muñoz, M.; Vey, J.; Eckert, C.; et al. Novel Autoantibody Signatures in Sera of Patients with Pancreatic Cancer, Chronic Pancreatitis and Autoimmune Pancreatitis: A Protein Microarray Profiling Approach. Int. J. Mol. Sci. 2020, 21, 2403. https://doi.org/10.3390/ijms21072403
Ghassem-Zadeh S, Hufnagel K, Bauer A, Frossard J-L, Yoshida M, Kutsumi H, Acha-Orbea H, Neulinger-Muñoz M, Vey J, Eckert C, et al. Novel Autoantibody Signatures in Sera of Patients with Pancreatic Cancer, Chronic Pancreatitis and Autoimmune Pancreatitis: A Protein Microarray Profiling Approach. International Journal of Molecular Sciences. 2020; 21(7):2403. https://doi.org/10.3390/ijms21072403
Chicago/Turabian StyleGhassem-Zadeh, Sahar, Katrin Hufnagel, Andrea Bauer, Jean-Louis Frossard, Masaru Yoshida, Hiromu Kutsumi, Hans Acha-Orbea, Matthias Neulinger-Muñoz, Johannes Vey, Christoph Eckert, and et al. 2020. "Novel Autoantibody Signatures in Sera of Patients with Pancreatic Cancer, Chronic Pancreatitis and Autoimmune Pancreatitis: A Protein Microarray Profiling Approach" International Journal of Molecular Sciences 21, no. 7: 2403. https://doi.org/10.3390/ijms21072403
APA StyleGhassem-Zadeh, S., Hufnagel, K., Bauer, A., Frossard, J.-L., Yoshida, M., Kutsumi, H., Acha-Orbea, H., Neulinger-Muñoz, M., Vey, J., Eckert, C., Strobel, O., Hoheisel, J. D., & Felix, K. (2020). Novel Autoantibody Signatures in Sera of Patients with Pancreatic Cancer, Chronic Pancreatitis and Autoimmune Pancreatitis: A Protein Microarray Profiling Approach. International Journal of Molecular Sciences, 21(7), 2403. https://doi.org/10.3390/ijms21072403