Interspecies Communication in Holobionts by Non-Coding RNA Exchange





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Concept of Cognitive-Based Evolution Beyond Post-Darwinism Theories
2. Holobionts in Cognitive Evolution: The Hologenome
3. The Non-Coding RNA Language for Cell-to-Cell Communications
3.1. Diversity of RNA Species in Transcriptomic Output
3.2. Conveyors for ncRNA Transport between Cells
3.3. Molecular Machinery for Understanding the External ncRNA Message
4. Cross-Kingdom Communication Mediated by ncRNAs in the Holobiont Context
4.1. Communication by ncRNAs Acting within Eukaryotic Cells in Holobionts
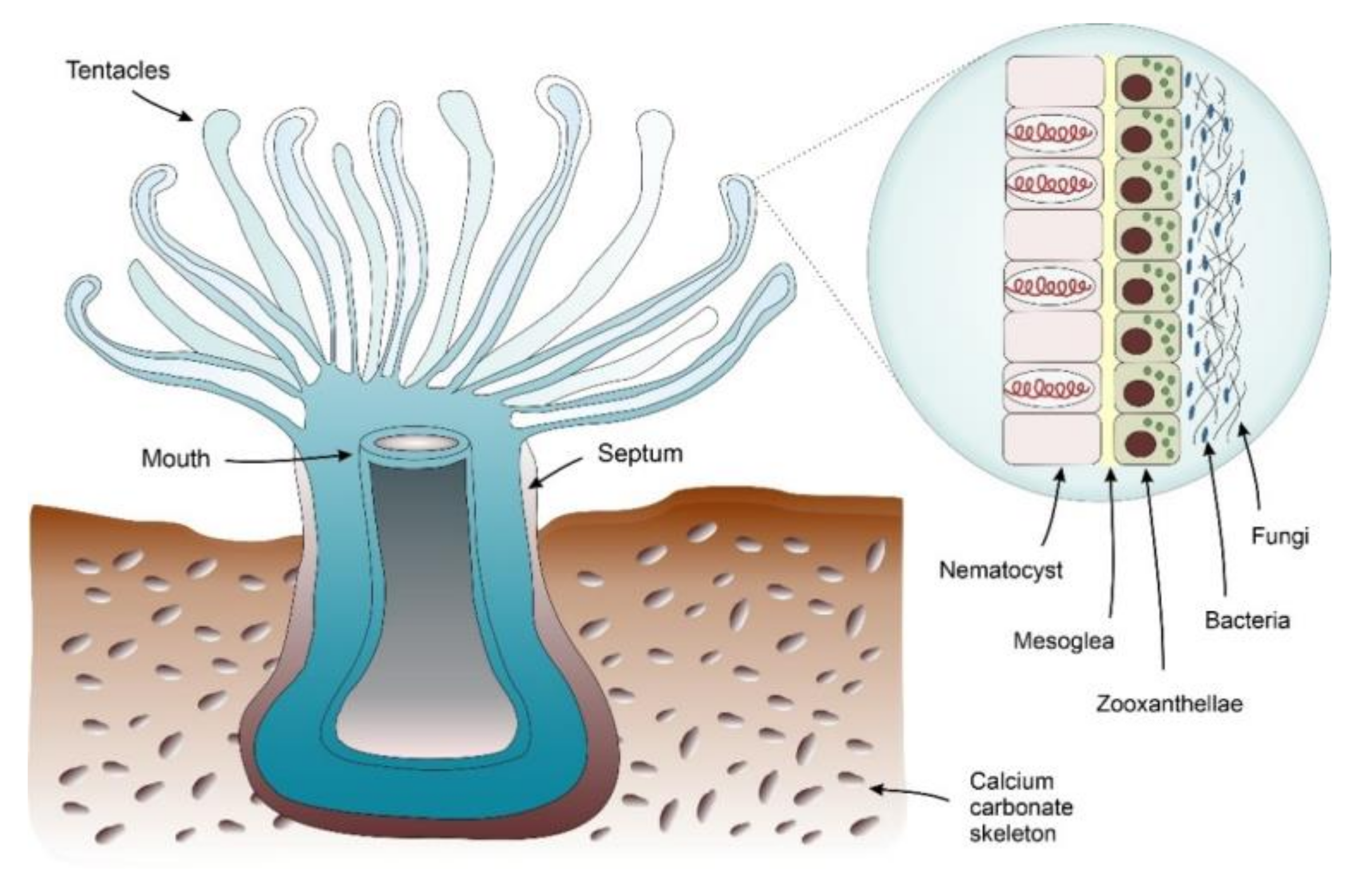
4.1.1. Corals
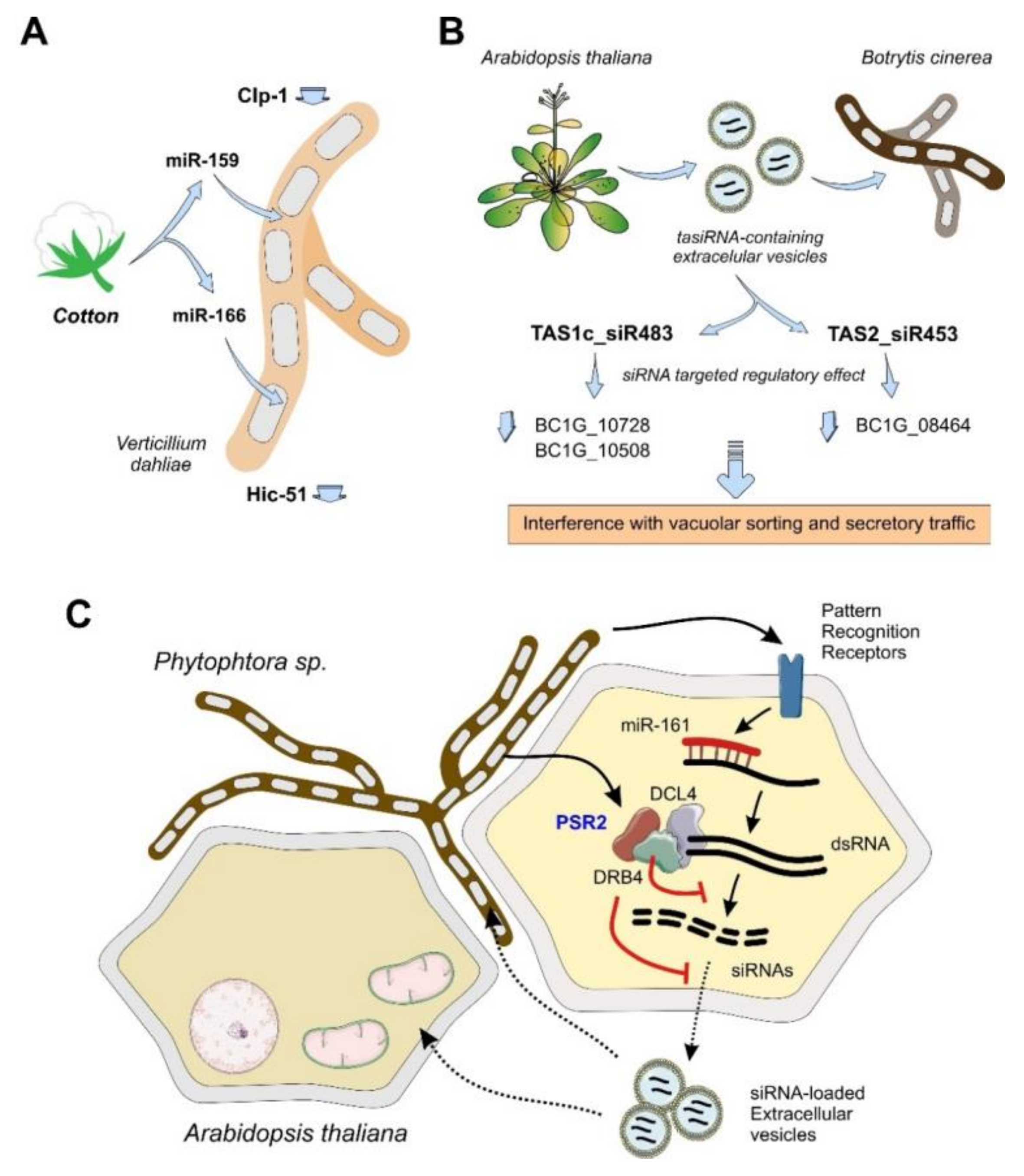
4.1.2. Plants
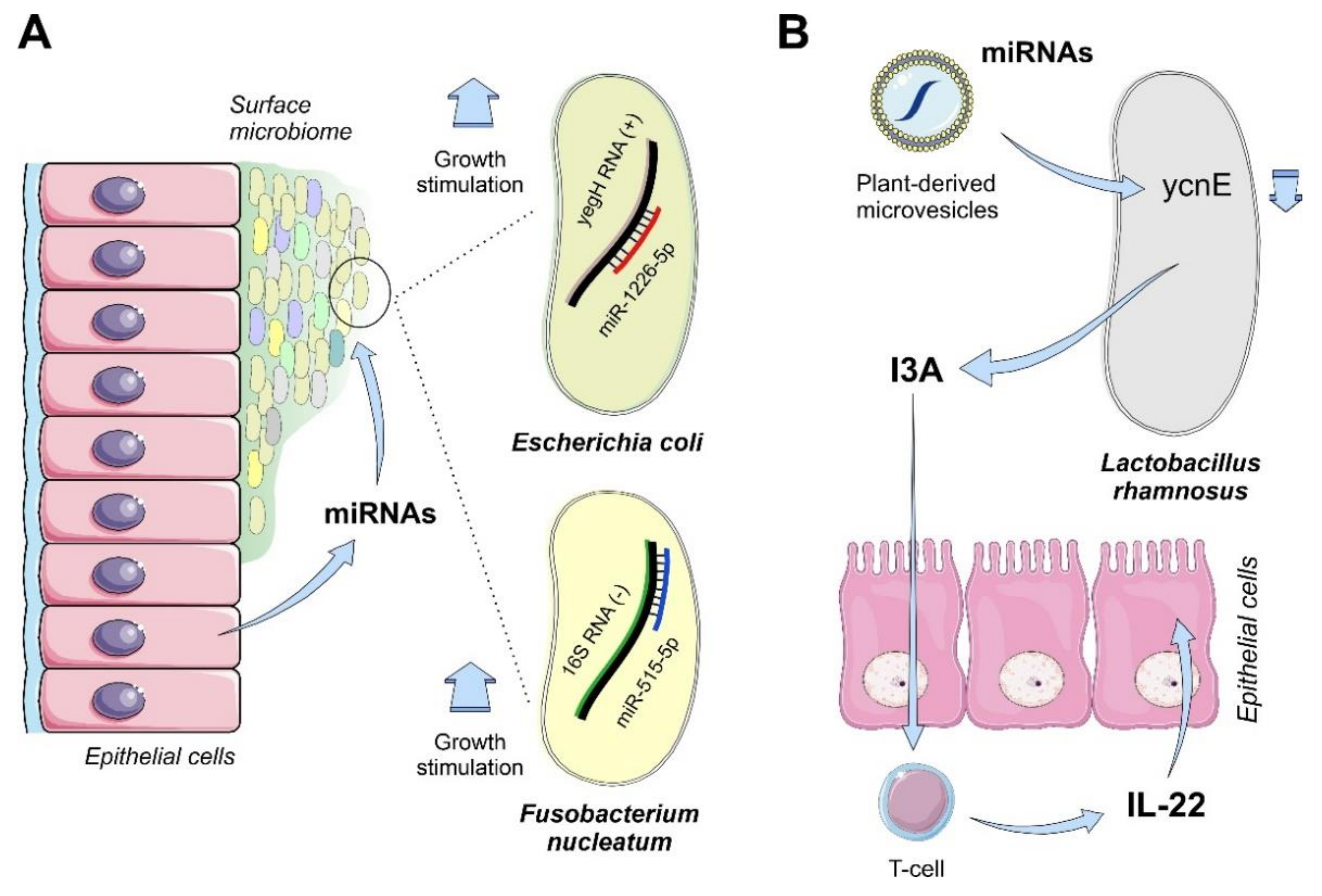
4.1.3. Mammals
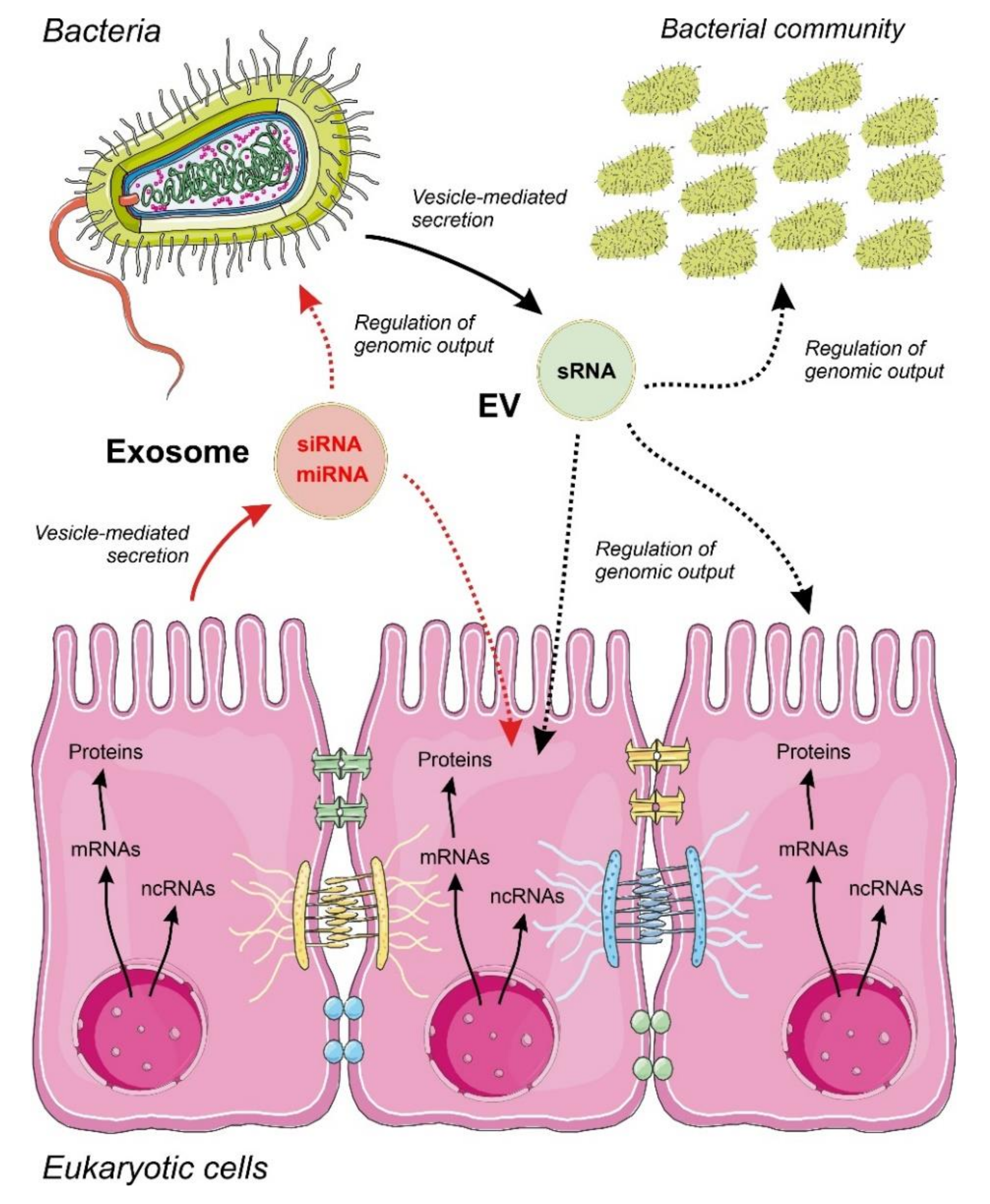
4.2. Communication by ncRNAs Acting within Bacteria in Multi-Species Consortia
5. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Noble, D. Evolution beyond neo-Darwinism: A new conceptual framework. J. Exp. Biol. 2015, 218 Pt 8, 1273. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, W.B., Jr.; Torday, J.S. Four domains: The fundamental unicell and Post-Darwinian Cognition-Based Evolution. Prog. Biophys. Mol. Biol. 2018, 140, 49–73. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.B., Jr. Biological information systems: Evolution as cognition-based information management. Prog. Biophys. Mol. Biol. 2018, 134, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A. Bacteria are small but not stupid: Cognition, natural genetic engineering and socio-bacteriology. Stud. Hist. Philos. Biol. Biomed. Sci. 2007, 38, 807–819. [Google Scholar] [CrossRef]
- Miller, W.B. Cognition, information fields and hologenomic entanglement: Evolution in light and shadow. Biology 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Mindell, D.P. Phylogenetic consequences of symbioses: Eukarya and Eubacteria are not monophyletic taxa. Biosystems 1992, 27, 53–62. [Google Scholar] [CrossRef]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef]
- Waldor, M.K.; Bordenstein, S.R.; Theis, K.R. Host biology in light of the microbiome: Ten principles of holobionts and hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar]
- Rosenberg, E.; Zilber-Rosenberg, I. The hologenome concept of evolution after 10 years. Microbiome 2018, 6, 78. [Google Scholar] [CrossRef]
- Morris, J.J. What is the hologenome concept of evolution? F1000Research 2018, 7, 1664. [Google Scholar] [CrossRef]
- Collens, A.; Kelley, E.; Katz, L.A. The concept of the hologenome, an epigenetic phenomenon, challenges aspects of the modern evolutionary synthesis. J. Exp. Zool. B Mol. Dev. Evol. 2019, 332, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Carrier, T.J.; Reitzel, A.M. The hologenome across environments and the implications of a host-associated microbial repertoire. Front. Microbiol. 2017, 8, 802. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Ganem, B. RNA world. Nature 1987, 328, 676. [Google Scholar] [CrossRef]
- Lozada-Chavez, I.; Stadler, P.F.; Prohaska, S.J. “Hypothesis for the modern RNA world”: A pervasive non-coding RNA-based genetic regulation is a prerequisite for the emergence of multicellular complexity. Orig. Life Evol. Biosph. 2011, 41, 587–607. [Google Scholar] [CrossRef]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Mattick, J.S. Pervasive transcription of the eukaryotic genome: Functional indices and conceptual implications. Brief. Funct. Genom. Proteom. 2009, 8, 407–423. [Google Scholar] [CrossRef]
- Lybecker, M.; Bilusic, I.; Raghavan, R. Pervasive transcription: Detecting functional RNAs in bacteria. Transcription 2014, 5, e944039. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Vaughn, I.W.; McManus, M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9, e1003569. [Google Scholar] [CrossRef]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, biology and functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Small regulatory RNAs in mammals. Hum. Mol. Genet. 2005, 14, R121–R132. [Google Scholar] [CrossRef]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Sarropoulos, I.; Marin, R.; Cardoso-Moreira, M.; Kaessmann, H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature 2019, 571, 510–514. [Google Scholar] [CrossRef]
- Huang, C.; Shan, G. What happens at or after transcription: Insights into circRNA biogenesis and function. Transcription 2015, 6, 61–64. [Google Scholar] [CrossRef]
- Tuck, A.C.; Natarajan, K.N.; Rice, G.M.; Borawski, J.; Mohn, F.; Rankova, A.; Flemr, M.; Wenger, A.; Nutiu, R.; Teichmann, S.; et al. Distinctive features of lincRNA gene expression suggest widespread RNA-independent functions. Life Sci. Alliance 2018, 1, e201800124. [Google Scholar] [CrossRef]
- Carrier, M.C.; Lalaouna, D.; Masse, E. Broadening the definition of bacterial small RNAs: Characteristics and mechanisms of action. Annu. Rev. Microbiol. 2018, 72, 141–161. [Google Scholar] [CrossRef]
- Nitzan, M.; Rehani, R.; Margalit, H. Integration of bacterial small RNAs in regulatory networks. Annu. Rev. Biophys. 2017, 46, 131–148. [Google Scholar] [CrossRef]
- Coleman, J.; Green, P.J.; Inouye, M. The use of RNAs complementary to specific mRNAs to regulate the expression of individual bacterial genes. Cell 1984, 37, 429–436. [Google Scholar] [CrossRef]
- Opdyke, J.A.; Fozo, E.M.; Hemm, M.R.; Storz, G. RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J. Mol. Biol. 2011, 406, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Deng, Z.; Yan, Y.; Zhang, H.; Lu, C.; Yang, Z.; Shang, L.; Huang, Y.; Lv, F.; Liu, Y.; et al. NfiR, a new regulatory noncoding RNA (ncRNA), is required in concert with the NfiS ncRNA for optimal expression of nitrogenase genes in pseudomonas stutzeri A1501. Appl. Environ. Microbiol. 2019, 85, e00762-19. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, S.C.; Hong, S.H.; Lee, H.J. Secretable small RNAs via outer membrane vesicles in periodontal pathogens. J. Dent. Res. 2017, 96, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.C.; Marchesi, J.R.; Mougel, C.; Selosse, M.A. Host-microbiota interactions: From holobiont theory to analysis. Microbiome 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, J.; Sun, Y.; Kwan, Y.H.; Wong, W.C.; Zhang, Y.; Xu, T.; Feng, D.; Zhang, Y.; Qiu, J.W.; et al. Genomic, transcriptomic, and proteomic insights into the symbiosis of deep-sea tubeworm holobionts. ISME J. 2020, 14, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Kalra, H.; Drummen, G.P.; Mathivanan, S. Focus on extracellular vesicles: Introducing the next small big thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Park, J.; Gho, Y.S. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin. Cell Dev. Biol. 2015, 40, 97–104. [Google Scholar] [CrossRef]
- Ahmadi Badi, S.; Moshiri, A.; Fateh, A.; Rahimi Jamnani, F.; Sarshar, M.; Vaziri, F.; Siadat, S.D. Microbiota-derived extracellular vesicles as new systemic regulators. Front. Microbiol. 2017, 8, 1610. [Google Scholar] [CrossRef]
- Lee, H.; Li, C.; Zhang, Y.; Zhang, D.; Otterbein, L.E.; Jin, Y. Caveolin-1 selectively regulates microRNA sorting into microvesicles after noxious stimuli. J. Exp. Med. 2019, 216, 2202–2220. [Google Scholar] [CrossRef]
- Temoche-Diaz, M.M.; Shurtleff, M.J.; Nottingham, R.M.; Yao, J.; Fadadu, R.P.; Lambowitz, A.M.; Schekman, R. Distinct mechanisms of microRNA sorting into cancer cell-derived extracellular vesicle subtypes. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. eLife 2016, 5, e19276. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; Baer, C.; Burdet, F.; Maderna, C.; Gilfillan, G.D.; Lyle, R.; Ibberson, M.; De Palma, M. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014, 8, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The host shapes the gut microbiota via fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef]
- Viennois, E.; Chassaing, B.; Tahsin, A.; Pujada, A.; Wang, L.; Gewirtz, A.T.; Merlin, D. Host-derived fecal microRNAs can indicate gut microbiota healthiness and ability to induce inflammation. Theranostics 2019, 9, 4542–4557. [Google Scholar] [CrossRef]
- Blenkiron, C.; Simonov, D.; Muthukaruppan, A.; Tsai, P.; Dauros, P.; Green, S.; Hong, J.; Print, C.G.; Swift, S.; Phillips, A.R. Uropathogenic Escherichia coli releases extracellular vesicles that are associated with RNA. PLoS ONE 2016, 11, e0160440. [Google Scholar] [CrossRef]
- Koeppen, K.; Hampton, T.H.; Jarek, M.; Scharfe, M.; Gerber, S.A.; Mielcarz, D.W.; Demers, E.G.; Dolben, E.L.; Hammond, J.H.; Hogan, D.A.; et al. A novel mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 2016, 12, e1005672. [Google Scholar] [CrossRef]
- Wang, M.; Weiberg, A.; Lin, F.-M.; Thomma, B.P.H.J.; Huang, H.-D.; Jin, H. Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat. Plants 2016, 2, 16151. [Google Scholar] [CrossRef]
- Carmell, M.A.; Xuan, Z.; Zhang, M.Q.; Hannon, G.J. The Argonaute family: Tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev. 2002, 16, 2733–2742. [Google Scholar] [CrossRef]
- Braun, L.; Cannella, D.; Ortet, P.; Barakat, M.; Sautel, C.F.; Kieffer, S.; Garin, J.; Bastien, O.; Voinnet, O.; Hakimi, M.A. A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog. 2010, 6, e1000920. [Google Scholar] [CrossRef]
- Nguyen, Q.; Iritani, A.; Ohkita, S.; Vu, B.V.; Yokoya, K.; Matsubara, A.; Ikeda, K.I.; Suzuki, N.; Nakayashiki, H. A fungal Argonaute interferes with RNA interference. Nucleic Acids Res. 2018, 46, 2495–2508. [Google Scholar] [CrossRef] [PubMed]
- Fatyol, K.; Ludman, M.; Burgyan, J. Functional dissection of a plant Argonaute. Nucleic Acids Res. 2016, 44, 1384–1397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, R.; Meyers, B.C.; Walbot, V. Evolution, functions, and mysteries of plant Argonaute proteins. Curr. Opin. Plant Biol. 2015, 27, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Shiohama, A.; Minoshima, S.; Shimizu, N. Identification of eight members of the Argonaute family in the human genome. Genomics 2003, 82, 323–330. [Google Scholar] [CrossRef]
- Hock, J.; Meister, G. The Argonaute protein family. Genome Biol. 2008, 9, 210. [Google Scholar] [CrossRef]
- Zhou, X.; Guo, H.; Chen, K.; Cheng, H.; Zhou, R. Identification, chromosomal mapping and conserved synteny of porcine Argonaute family of genes. Genetica 2010, 138, 805–812. [Google Scholar] [CrossRef]
- Eulalio, A.; Huntzinger, E.; Nishihara, T.; Rehwinkel, J.; Fauser, M.; Izaurralde, E. Deadenylation is a widespread effect of miRNA regulation. RNA 2009, 15, 21–32. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Wang, H.; Hu, P.; Hamby, R.; Jin, H. Small RNAs—Big players in plant-microbe interactions. Cell Host Microbe 2019, 26, 173–182. [Google Scholar] [CrossRef]
- Buck, A.H.; Coakley, G.; Simbari, F.; McSorley, H.J.; Quintana, J.F.; Le Bihan, T.; Kumar, S.; Abreu-Goodger, C.; Lear, M.; Harcus, Y.; et al. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat. Commun. 2014, 5, 5488. [Google Scholar] [CrossRef]
- Shahid, S.; Kim, G.; Johnson, N.R.; Wafula, E.; Wang, F.; Coruh, C.; Bernal-Galeano, V.; Phifer, T.; dePamphilis, C.W.; Westwood, J.H.; et al. MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature 2018, 553, 82–85. [Google Scholar] [CrossRef]
- Zhang, A.; Wassarman, K.M.; Rosenow, C.; Tjaden, B.C.; Storz, G.; Gottesman, S. Global analysis of small RNA and mRNA targets of Hfq. Mol. Microbiol. 2003, 50, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.; Uno, K.; Morita, T.; Aiba, H. RNA, but not protein partners, is directly responsible for translational silencing by a bacterial Hfq-binding small RNA. Proc. Natl. Acad. Sci. USA 2008, 105, 10332–10337. [Google Scholar] [CrossRef] [PubMed]
- Aiba, H. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 2007, 10, 134–139. [Google Scholar] [CrossRef] [PubMed]
- De Lay, N.; Schu, D.J.; Gottesman, S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013, 288, 7996–8003. [Google Scholar] [CrossRef]
- Oliva, G.; Sahr, T.; Rolando, M.; Knoth, M.; Buchrieser, C. A Unique cis-encoded small noncoding RNA is regulating legionella pneumophila Hfq expression in a life cycle-dependent manner. MBio 2017, 8, e02182-16. [Google Scholar] [CrossRef]
- Schiano, C.A.; Bellows, L.E.; Lathem, W.W. The small RNA chaperone Hfq is required for the virulence of Yersinia pseudotuberculosis. Infect. Immun. 2010, 78, 2034–2044. [Google Scholar] [CrossRef]
- Islam, W.; Islam, S.U.; Qasim, M.; Wang, L. Host-Pathogen interactions modulated by small RNAs. RNA Biol. 2017, 14, 891–904. [Google Scholar] [CrossRef]
- Shah, N.; Tang, H.; Doak, T.G.; Ye, Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. In Pacific Symposium on Biocomputing; World Scientific Publishing Company: Singapore, 2011; pp. 165–176. [Google Scholar]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar] [CrossRef]
- Nowicki, E.M.; Shroff, R.; Singleton, J.A.; Renaud, D.E.; Wallace, D.; Drury, J.; Zirnheld, J.; Colleti, B.; Ellington, A.D.; Lamont, R.J.; et al. Microbiota and metatranscriptome changes accompanying the onset of gingivitis. MBio 2018, 9, e00575-18. [Google Scholar] [CrossRef]
- Peterson, B.F.; Scharf, M.E. Metatranscriptome analysis reveals bacterial symbiont contributions to lower termite physiology and potential immune functions. BMC Genom. 2016, 17, 772. [Google Scholar] [CrossRef]
- Daniels, C.A.; Baumgarten, S.; Yum, L.K.; Michell, C.T.; Bayer, T.; Arif, C.; Roder, C.; Weil, E.; Voolstra, C.R. Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Front. Mar. Sci. 2015, 2, 62. [Google Scholar] [CrossRef]
- Shakya, M.; Lo, C.C.; Chain, P.S.G. Advances and challenges in metatranscriptomic analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Greer, R.; Dong, X.; Morgun, A.; Shulzhenko, N. Investigating a holobiont: Microbiota perturbations and transkingdom networks. Gut Microbes 2016, 7, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Krediet, C.J.; Ritchie, K.B.; Paul, V.J.; Teplitski, M. Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proc. R. Soc. B Biol. Sci. 2013, 280, 20122328. [Google Scholar] [CrossRef]
- Pinzon, J.H.; Kamel, B.; Burge, C.A.; Harvell, C.D.; Medina, M.; Weil, E.; Mydlarz, L.D. Whole transcriptome analysis reveals changes in expression of immune-related genes during and after bleaching in a reef-building coral. R. Soc. Open Sci. 2015, 2, 140214. [Google Scholar] [CrossRef]
- Burge, C.A.; Mouchka, M.E.; Harvell, C.D.; Roberts, S. Immune response of the Caribbean sea fan, Gorgonia ventalina, exposed to an Aplanochytrium parasite as revealed by transcriptome sequencing. Front. Physiol. 2013, 4, 180. [Google Scholar] [CrossRef]
- Libro, S.; Kaluziak, S.T.; Vollmer, S.V. RNA-seq profiles of immune related genes in the staghorn coral Acropora cervicornis infected with white band disease. PLoS ONE 2013, 8, e81821. [Google Scholar] [CrossRef]
- van de Water, J.; Chaib De Mares, M.; Dixon, G.B.; Raina, J.B.; Willis, B.L.; Bourne, D.G.; van Oppen, M.J.H. Antimicrobial and stress responses to increased temperature and bacterial pathogen challenge in the holobiont of a reef-building coral. Mol. Ecol. 2018, 27, 1065–1080. [Google Scholar] [CrossRef]
- Hernandez-Agreda, A.; Gates, R.D.; Ainsworth, T.D. Defining the core microbiome in corals’ microbial soup. Trends Microbiol. 2017, 25, 125–140. [Google Scholar] [CrossRef]
- Putnam, H.M.; Barott, K.L.; Ainsworth, T.D.; Gates, R.D. The vulnerability and resilience of reef-building corals. Curr. Biol. 2017, 27, R528–R540. [Google Scholar] [CrossRef]
- Reshef, L.; Koren, O.; Loya, Y.; Zilber-Rosenberg, I.; Rosenberg, E. The coral probiotic hypothesis. Environ. Microbiol. 2006, 8, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Golberg, K.; Pavlov, V.; Marks, R.S.; Kushmaro, A. Coral-associated bacteria, quorum sensing disrupters, and the regulation of biofouling. Biofouling 2013, 29, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Pollock, F.J.; McMinds, R.; Smith, S.; Bourne, D.G.; Willis, B.L.; Medina, M.; Thurber, R.V.; Zaneveld, J.R. Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 2018, 9, 4921. [Google Scholar] [CrossRef] [PubMed]
- Soler-Hurtado, M.M.; Sandoval-Sierra, J.V.; Machordom, A.; Dieguez-Uribeondo, J. Aspergillus sydowii and other potential fungal pathogens in gorgonian octocorals of the Ecuadorian pacific. PLoS ONE 2016, 11, e0165992. [Google Scholar] [CrossRef] [PubMed]
- Planes, S.; Allemand, D.; Agostini, S.; Banaigs, B.; Boissin, E.; Boss, E.; Bourdin, G.; Bowler, C.; Douville, E.; Flores, J.M.; et al. The Tara Pacific expedition-A pan-ecosystemic approach of the “-omics” complexity of coral reef holobionts across the Pacific Ocean. PLoS Biol. 2019, 17, e3000483. [Google Scholar] [CrossRef]
- Frazier, M.; Helmkampf, M.; Bellinger, M.R.; Geib, S.M.; Takabayashi, M. De novo metatranscriptome assembly and coral gene expression profile of Montipora capitata with growth anomaly. BMC Genom. 2017, 18, 710. [Google Scholar] [CrossRef]
- Kaniewska, P.; Chan, C.K.; Kline, D.; Ling, E.Y.; Rosic, N.; Edwards, D.; Hoegh-Guldberg, O.; Dove, S. Transcriptomic changes in coral holobionts provide insights into physiological challenges of future climate and ocean change. PLoS ONE 2015, 10, e0139223. [Google Scholar] [CrossRef]
- Liew, Y.J.; Aranda, M.; Carr, A.; Baumgarten, S.; Zoccola, D.; Tambutte, S.; Allemand, D.; Micklem, G.; Voolstra, C.R. Identification of microRNAs in the coral Stylophora pistillata. PLoS ONE 2014, 9, e91101. [Google Scholar] [CrossRef]
- Gajigan, A.P.; Conaco, C. A microRNA regulates the response of corals to thermal stress. Mol. Ecol. 2017, 26, 3472–3483. [Google Scholar] [CrossRef]
- Huang, C.; Morlighem, J.R.L.; Cai, J.; Liao, Q.; Perez, C.D.; Gomes, P.B.; Guo, M.; Radis-Baptista, G.; Lee, S.M. Identification of long non-coding RNAs in two anthozoan species and their possible implications for coral bleaching. Sci. Rep. 2017, 7, 5333. [Google Scholar] [CrossRef]
- Baumgarten, S.; Cziesielski, M.J.; Thomas, L.; Michell, C.T.; Esherick, L.Y.; Pringle, J.R.; Aranda, M.; Voolstra, C.R. Evidence for miRNA-mediated modulation of the host transcriptome in cnidarian-dinoflagellate symbiosis. Mol. Ecol. 2018, 27, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, T.; Deng, Z.; Fu, H.; Lin, W. Janthinolide A-B, two new 2,5-piperazinedione derivatives from the endophytic Penicillium janthinellum isolated from the soft coral Dendronephthya sp. Pharmazie 2006, 61, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhou, Y.; Chen, X. New insight into inter-kingdom communication: Horizontal transfer of mobile small RNAs. Front. Microbiol. 2017, 8, 768. [Google Scholar] [CrossRef]
- Hacquard, S. Disentangling the factors shaping microbiota composition across the plant holobiont. New Phytol. 2016, 209, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Hiltner, L. Uber neuere Erfahrungen und Probleme auf dem Gebiete der Bodenbakteriologie unter besonderden berucksichtigung und Brache. Arb. Dtsch. Landwirtsch. Ges. 1904, 98, 59–78. [Google Scholar]
- Shakya, M.; Gottel, N.; Castro, H.; Yang, Z.K.; Gunter, L.; Labbe, J.; Muchero, W.; Bonito, G.; Vilgalys, R.; Tuskan, G.; et al. A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature Populus deltoides trees. PLoS ONE 2013, 8, e76382. [Google Scholar] [CrossRef]
- Maignien, L.; DeForce, E.A.; Chafee, M.E.; Eren, A.M.; Simmons, S.L. Ecological succession and stochastic variation in the assembly of Arabidopsis thaliana phyllosphere communities. MBio 2014, 5, e00682-13. [Google Scholar] [CrossRef]
- Gonzalez, E.; Pitre, F.E.; Page, A.P.; Marleau, J.; Guidi Nissim, W.; St-Arnaud, M.; Labrecque, M.; Joly, S.; Yergeau, E.; Brereton, N.J.B. Trees, fungi and bacteria: Tripartite metatranscriptomics of a root microbiome responding to soil contamination. Microbiome 2018, 6, 53. [Google Scholar] [CrossRef]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y.; et al. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E4284–E4293. [Google Scholar] [CrossRef]
- Rodriguez, P.A.; Rothballer, M.; Chowdhury, S.P.; Nussbaumer, T.; Gutjahr, C.; Falter-Braun, P. Systems biology of plant-microbiome interactions. Mol. Plant 2019, 12, 804–821. [Google Scholar] [CrossRef]
- Glazebrook, J.; Roby, D. Plant biotic interactions: From conflict to collaboration. Plant J. 2018, 93, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, K.; Xie, X.; Kim, H.I.; Kisugi, T.; Nomura, T.; Sekimoto, H.; Yokota, T.; Yoneyama, K. How do nitrogen and phosphorus deficiencies affect strigolactone production and exudation? Planta 2012, 235, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, K.; Xie, X.; Kisugi, T.; Nomura, T.; Yoneyama, K. Nitrogen and phosphorus fertilization negatively affects strigolactone production and exudation in sorghum. Planta 2013, 238, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Leitao, A.L.; Enguita, F.J. Gibberellins in Penicillium strains: Challenges for endophyte-plant host interactions under salinity stress. Microbiol. Res. 2016, 183, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, C.; Li, S.; Peng, M. Long noncoding RNAs that respond to Fusarium oxysporum infection in ‘Cavendish’ banana (Musa acuminata). Sci. Rep. 2017, 7, 16939. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Xu, Y.; Huang, D.; Ashraf, M.A.; Li, J.; Hu, W.; Jin, Z.; Zeng, C.; Tang, F.; Xu, B.; et al. Identification and characterization of miRNA169 family members in banana (Musa acuminata L.) that respond to fusarium oxysporum f. sp. cubense infection in banana cultivars. PeerJ 2018, 6, e6209. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.L.; Zhao, J.H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef]
- Jiao, J.; Peng, D. Wheat microRNA1023 suppresses invasion of Fusarium graminearum via targeting and silencing FGSG_03101. J. Plant Interact. 2018, 13, 514–521. [Google Scholar] [CrossRef]
- Gabriel, A.F.; Costa, M.C.; Enguita, F.J.; Leitão, A.L. Si vis pacem para bellum: A prospective in silico analysis of miRNA-based plant defenses against fungal infections. Plant Sci. 2019, 288, 110241. [Google Scholar] [CrossRef]
- Cai, Q.; Qiao, L.; Wang, M.; He, B.; Lin, F.M.; Palmquist, J.; Huang, S.D.; Jin, H. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 2018, 360, 1126–1129. [Google Scholar] [CrossRef]
- Hou, Y.; Zhai, Y.; Feng, L.; Karimi, H.Z.; Rutter, B.D.; Zeng, L.; Choi, D.S.; Zhang, B.; Gu, W.; Chen, X.; et al. A phytophthora effector suppresses trans-kingdom RNAi to promote disease susceptibility. Cell Host Microbe 2019, 25, 153.e5–165.e5. [Google Scholar] [CrossRef] [PubMed]
- de Felippes, F.F.; Marchais, A.; Sarazin, A.; Oberlin, S.; Voinnet, O. A single miR390 targeting event is sufficient for triggering TAS3-tasiRNA biogenesis in Arabidopsis. Nucleic Acids Res. 2017, 45, 5539–5554. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lin, L.; Lai, R.; Liu, W.; Chen, Y.; Zhang, Z.; XuHan, X.; Lai, Z. MicroRNA390-directed TAS3 cleavage leads to the production of tasiRNA-ARF3/4 during somatic embryogenesis in Dimocarpus longan lour. Front. Plant Sci. 2015, 6, 1119. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, S.; Tabara, M.; Nishibori, Y.; Moriyama, H.; Fukuhara, T. Long DCL4-substrate dsRNAs efficiently induce RNA interference in plant cells. Sci. Rep. 2019, 9, 6920. [Google Scholar] [CrossRef] [PubMed]
- Chiliveri, S.C.; Aute, R.; Rai, U.; Deshmukh, M.V. DRB4 dsRBD1 drives dsRNA recognition in Arabidopsis thaliana tasi/siRNA pathway. Nucleic Acids Res. 2017, 45, 8551–8563. [Google Scholar] [CrossRef]
- Ellegaard, K.M.; Engel, P. Beyond 16S rRNA community profiling: Intra-species diversity in the gut microbiota. Front. Microbiol. 2016, 7, 1475. [Google Scholar] [CrossRef]
- Al-Asmakh, M.; Zadjali, F. Use of germ-free animal models in microbiota-related research. J. Microbiol. Biotechnol. 2015, 25, 1583–1588. [Google Scholar] [CrossRef]
- Grover, M.; Kashyap, P.C. Germ-free mice as a model to study effect of gut microbiota on host physiology. Neurogastroenterol. Motil. 2014, 26, 745–748. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef]
- Gallo, R.L.; Hultsch, T.; Farnaes, L. Recognizing that the microbiome is part of the human immune system will advance treatment of both cancer and infections. J. Am. Acad. Dermatol. 2016, 74, 772–774. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mutlu, E.A.; Keshavarzian, A.; Losurdo, J.; Swanson, G.; Siewe, B.; Forsyth, C.; French, A.; Demarais, P.; Sun, Y.; Koenig, L.; et al. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 2014, 10, e1003829. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.E.; Griffith, T.S. A wild microbiome improves mouse modeling of the human immune response. Lab. Anim. N. Y. 2019, 48, 337–338. [Google Scholar] [CrossRef]
- Ibiza, S.; Garcia-Cassani, B.; Ribeiro, H.; Carvalho, T.; Almeida, L.; Marques, R.; Misic, A.M.; Bartow-McKenney, C.; Larson, D.M.; Pavan, W.J.; et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 2016, 535, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Godinho-Silva, C.; Domingues, R.G.; Rendas, M.; Raposo, B.; Ribeiro, H.; da Silva, J.A.; Vieira, A.; Costa, R.M.; Barbosa-Morais, N.L.; Carvalho, T.; et al. Light-entrained and brain-tuned circadian circuits regulate ILC3s and gut homeostasis. Nature 2019, 574, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, S.; Ferrero, G.; Gallo, G.; Francavilla, A.; Clerico, G.; Realis Luc, A.; Manghi, P.; Thomas, A.M.; Vineis, P.; Segata, N.; et al. Altered fecal small RNA profiles in colorectal cancer reflect gut microbiome composition in stool samples. mSystems 2019, 4, e00289-19. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Sears, C.L. Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: Contributions to colorectal cancer development. Genome Med. 2019, 11, 11. [Google Scholar] [CrossRef]
- Iacob, S.; Iacob, D.G. Infectious threats, the intestinal barrier, and its trojan horse: Dysbiosis. Front. Microbiol. 2019, 10, 1676. [Google Scholar] [CrossRef]
- Zhou, X.; Li, X.; Wu, M. miRNAs reshape immunity and inflammatory responses in bacterial infection. Signal Transduct. Target. Ther. 2018, 3, 14. [Google Scholar] [CrossRef]
- Tatematsu, M.; Funami, K.; Seya, T.; Matsumoto, M. Extracellular RNA sensing by pattern recognition receptors. J. Innate Immun. 2018, 10, 398–406. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Zhang, A.R.; Xu, M.; Lou, J.; Qiu, W.Q. TLR-4/miRNA-32-5p/FSTL1 signaling regulates mycobacterial survival and inflammatory responses in Mycobacterium tuberculosis-infected macrophages. Exp. Cell Res. 2017, 352, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, X.; Xiao, B.; Zhu, E.D.; Li, B.S.; Liu, Z.; Tang, B.; Zou, Q.M.; Liang, H.P.; Mao, X.H. H. pylori related proinflammatory cytokines contribute to the induction of miR-146a in human gastric epithelial cells. Mol. Biol. Rep. 2012, 39, 4655–4661. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Plaza, J.J. Small RNAs in cell-to-cell communications during bacterial infection. FEMS Microbiol. Lett. 2018, 365, fny024. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zhao, C.; Zhang, T.; Liang, H.; Wang, X.M.; Pan, Y.; Chen, X.; Zhao, Q.; Li, D.; Liu, F.; et al. Salmonella produce microRNA-like RNA fragment Sal-1 in the infected cells to facilitate intracellular survival. Sci. Rep. 2017, 7, 2392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhou, Z.; Zhang, T.; Liu, F.; Zhang, C.Y.; Zen, K.; Gu, H. Salmonella small RNA fragment Sal-1 facilitates bacterial survival in infected cells via suppressing iNOS induction in a microRNA manner. Sci. Rep. 2017, 7, 16979. [Google Scholar] [CrossRef]
- Babatunde, K.A.; Mbagwu, S.; Hernandez-Castaneda, M.A.; Adapa, S.R.; Walch, M.; Filgueira, L.; Falquet, L.; Jiang, R.H.Y.; Ghiran, I.; Mantel, P.Y. Malaria infected red blood cells release small regulatory RNAs through extracellular vesicles. Sci. Rep. 2018, 8, 884. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, L.; Li, J.; Wang, L.; Wu, Z.; Sun, X. Extracellular vesicle-mediated communication within host-parasite interactions. Front. Immunol. 2018, 9, 3066. [Google Scholar] [CrossRef]
- Dandewad, V.; Vindu, A.; Joseph, J.; Seshadri, V. Import of human miRNA-RISC complex into Plasmodium falciparum and regulation of the parasite gene expression. J. Biosci. 2019, 44, 50. [Google Scholar] [CrossRef]
- Bayer-Santos, E.; Marini, M.M.; da Silveira, J.F. Non-coding RNAs in host-pathogen interactions: Subversion of mammalian cell functions by protozoan parasites. Front. Microbiol. 2017, 8, 474. [Google Scholar] [CrossRef]
- Judice, C.C.; Bourgard, C.; Kayano, A.C.; Albrecht, L.; Costa, F.T. MicroRNAs in the host-apicomplexan parasites interactions: A review of immunopathological aspects. Front. Cell. Infect. Microbiol. 2016, 6, 5. [Google Scholar] [CrossRef]
- Arora, N.; Tripathi, S.; Singh, A.K.; Mondal, P.; Mishra, A.; Prasad, A. Micromanagement of immune system: Role of miRNAs in helminthic infections. Front. Microbiol. 2017, 8, 586. [Google Scholar] [CrossRef]
- Waters, L.S.; Storz, G. Regulatory RNAs in bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Pinho, F.G.; Frampton, A.E.; Nunes, J.; Krell, J.; Alshaker, H.; Jacob, J.; Pellegrino, L.; Roca-Alonso, L.; de Giorgio, A.; Harding, V.; et al. Downregulation of microRNA-515-5p by the estrogen receptor modulates sphingosine kinase 1 and breast cancer cell proliferation. Cancer Res. 2013, 73, 5936–5948. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Castellano, L.; Munro, C.E.; Hu, Y.; Mauri, F.; Krell, J.; Lara, R.; Pinho, F.G.; Choudhury, T.; Frampton, A.E.; et al. miR-515-5p controls cancer cell migration through MARK4 regulation. EMBO Rep. 2016, 17, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Ren, Y.; Sayed, M.; Hu, X.; Lei, C.; Kumar, A.; Hutchins, E.; Mu, J.; Deng, Z.; Luo, C.; et al. Plant-derived exosomal MicroRNAs shape the gut microbiota. Cell Host Microbe 2018, 24, 637–652. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse microbiota models: Comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front. Physiol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- O’Brien, P.A.; Webster, N.S.; Miller, D.J.; Bourne, D.G. Host-microbe coevolution: Applying evidence from model systems to complex marine invertebrate holobionts. MBio 2019, 10, e02241-18. [Google Scholar]
- Daharsh, L.; Zhang, J.; Ramer-Tait, A.; Li, Q. A double humanized BLT-mice model featuring a stable human-like gut microbiome and human immune system. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Gezsi, A.; Kovacs, A.; Visnovitz, T.; Buzas, E.I. Systems biology approaches to investigating the roles of extracellular vesicles in human diseases. Exp. Mol. Med. 2019, 51, 33. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitão, A.L.; Costa, M.C.; Gabriel, A.F.; Enguita, F.J. Interspecies Communication in Holobionts by Non-Coding RNA Exchange. Int. J. Mol. Sci. 2020, 21, 2333. https://doi.org/10.3390/ijms21072333
Leitão AL, Costa MC, Gabriel AF, Enguita FJ. Interspecies Communication in Holobionts by Non-Coding RNA Exchange. International Journal of Molecular Sciences. 2020; 21(7):2333. https://doi.org/10.3390/ijms21072333
Chicago/Turabian StyleLeitão, Ana Lúcia, Marina C. Costa, André F. Gabriel, and Francisco J. Enguita. 2020. "Interspecies Communication in Holobionts by Non-Coding RNA Exchange" International Journal of Molecular Sciences 21, no. 7: 2333. https://doi.org/10.3390/ijms21072333
APA StyleLeitão, A. L., Costa, M. C., Gabriel, A. F., & Enguita, F. J. (2020). Interspecies Communication in Holobionts by Non-Coding RNA Exchange. International Journal of Molecular Sciences, 21(7), 2333. https://doi.org/10.3390/ijms21072333