Upregulation of CASP9 through NF-κB and Its Target MiR-1276 Contributed to TNFα-Promoted Apoptosis of Cancer Cells Induced by Doxorubicin


Abstract
1. Introduction
2. Results
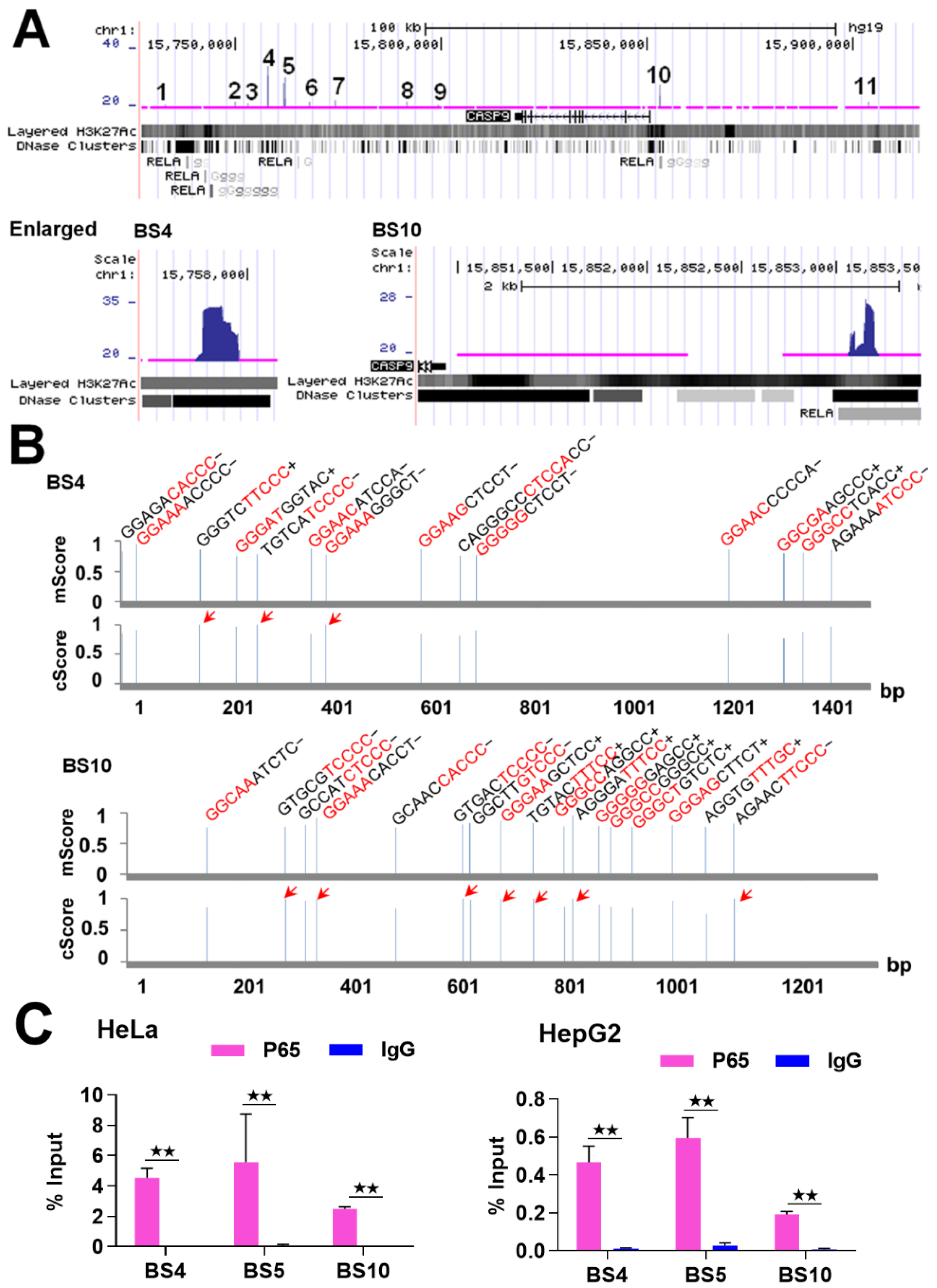
2.1. NF-κB Binds to CASP9
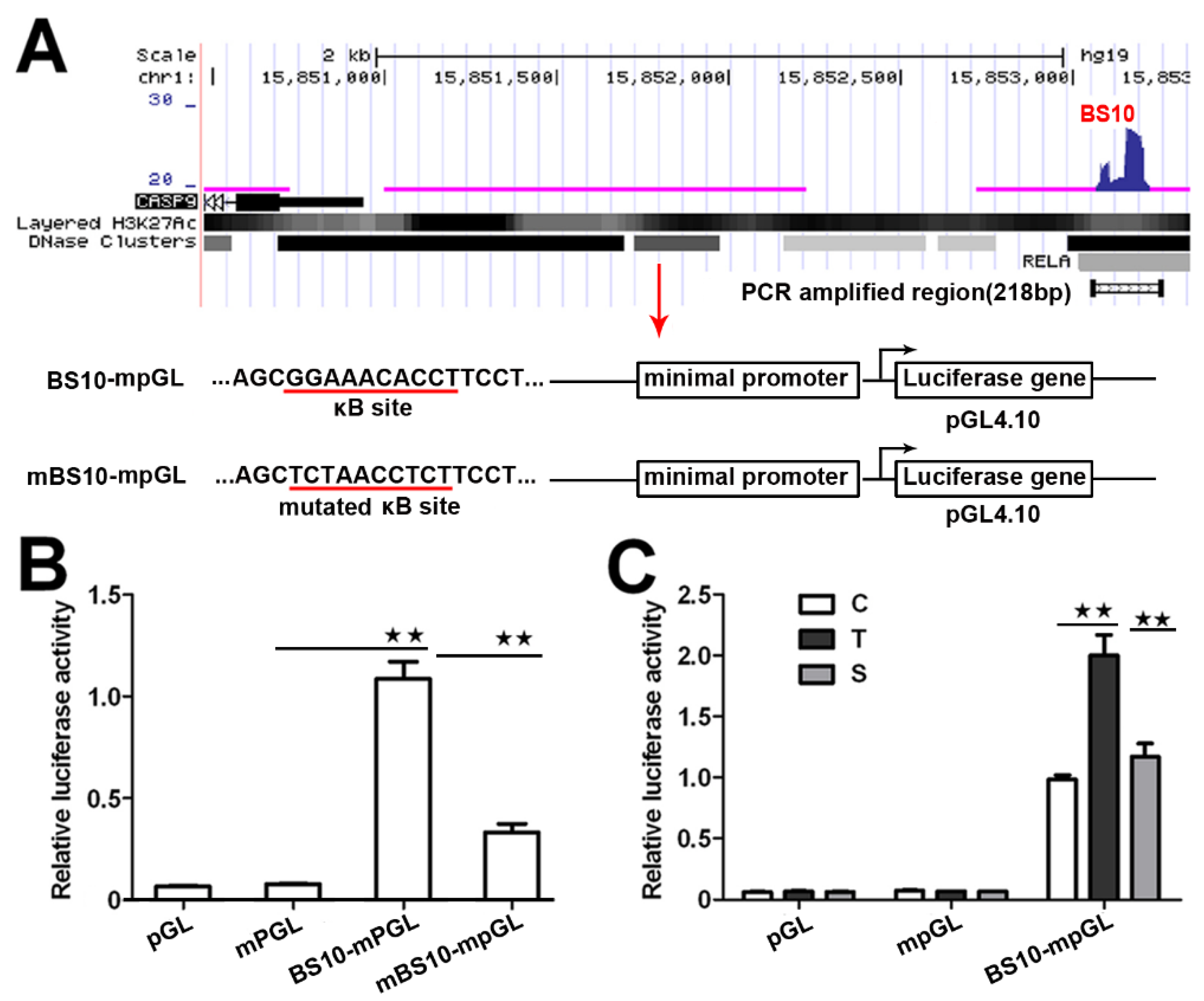
2.2. NF-κB Directly Regulates CASP9 Expression
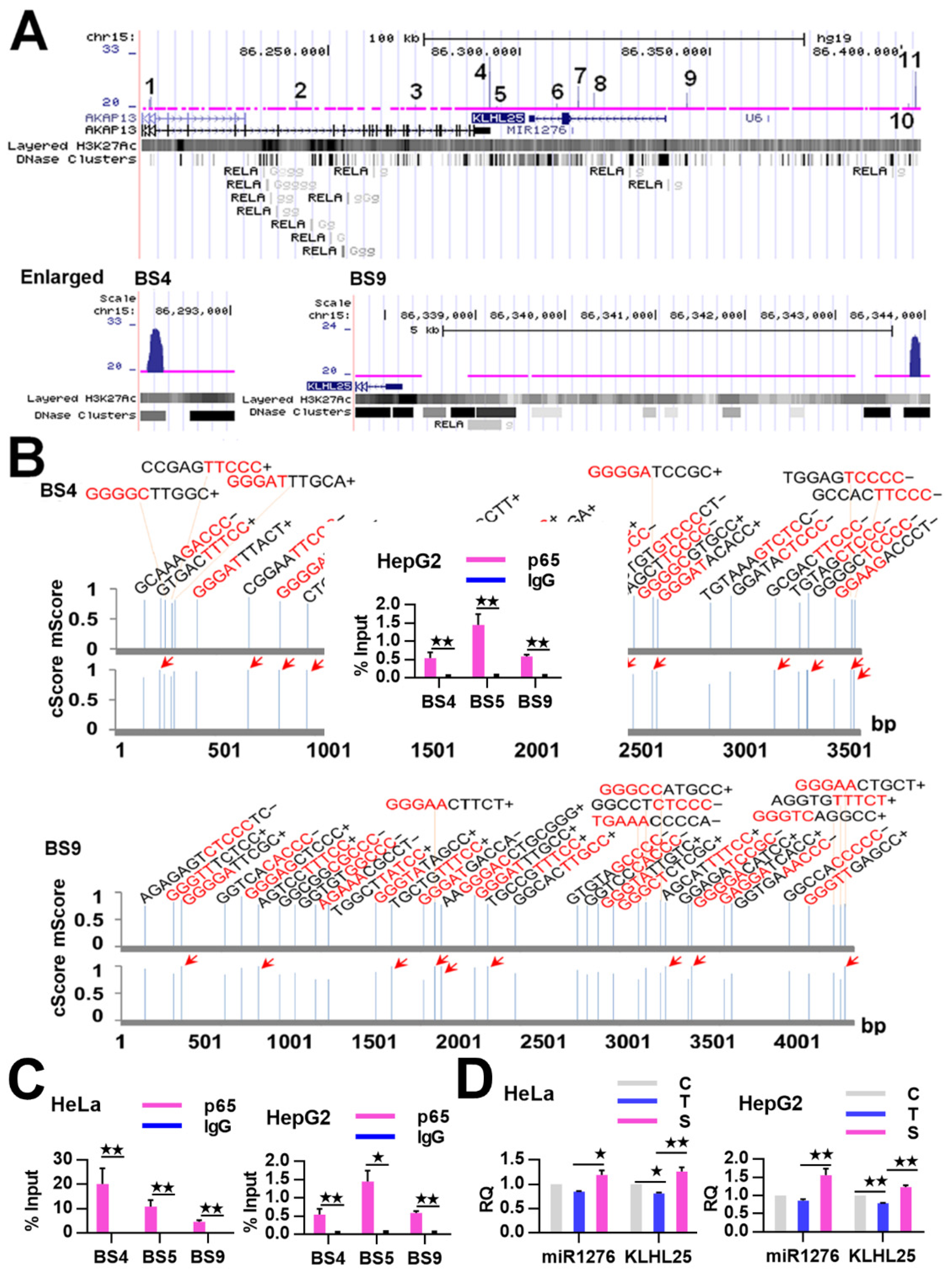
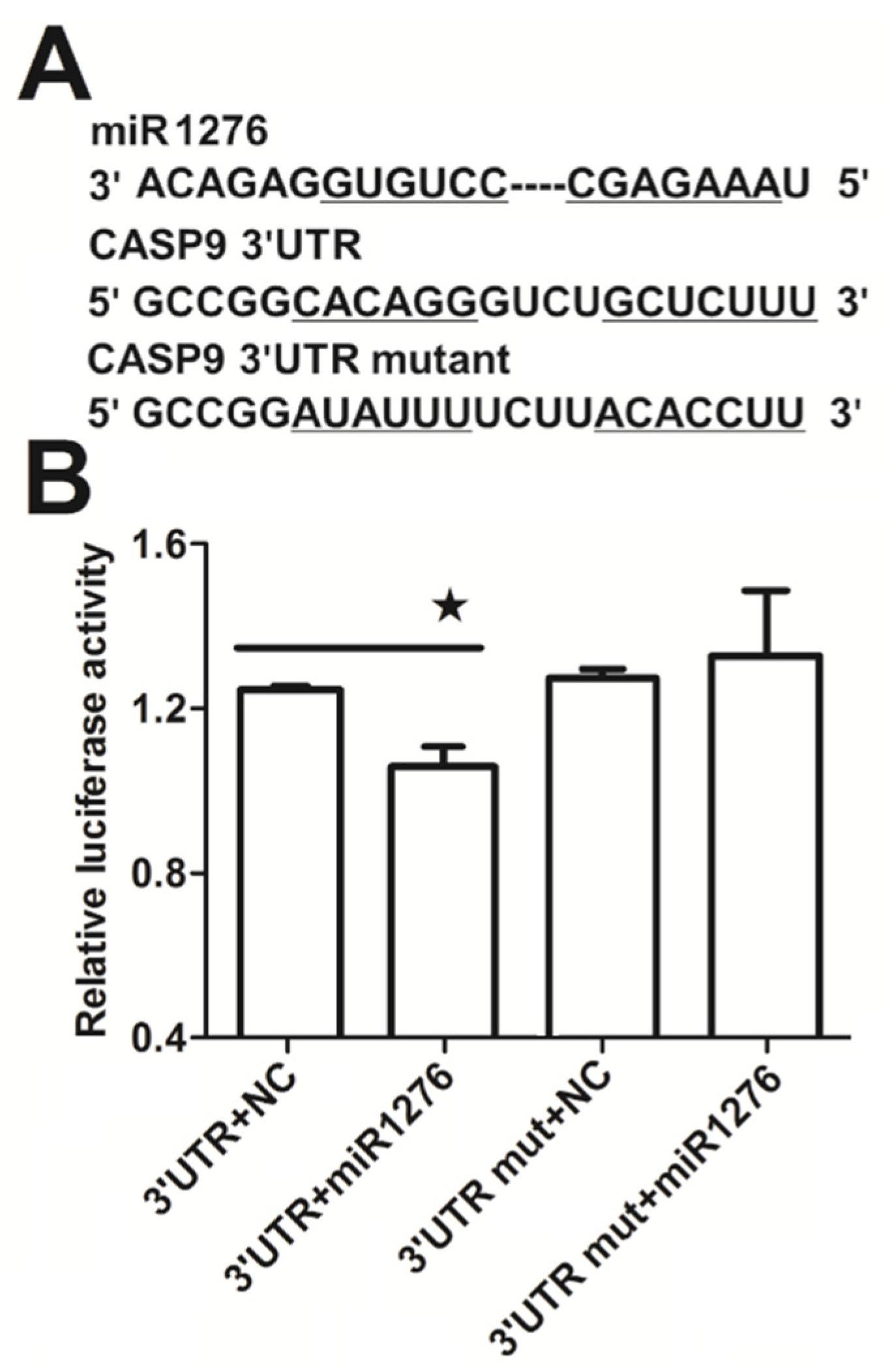
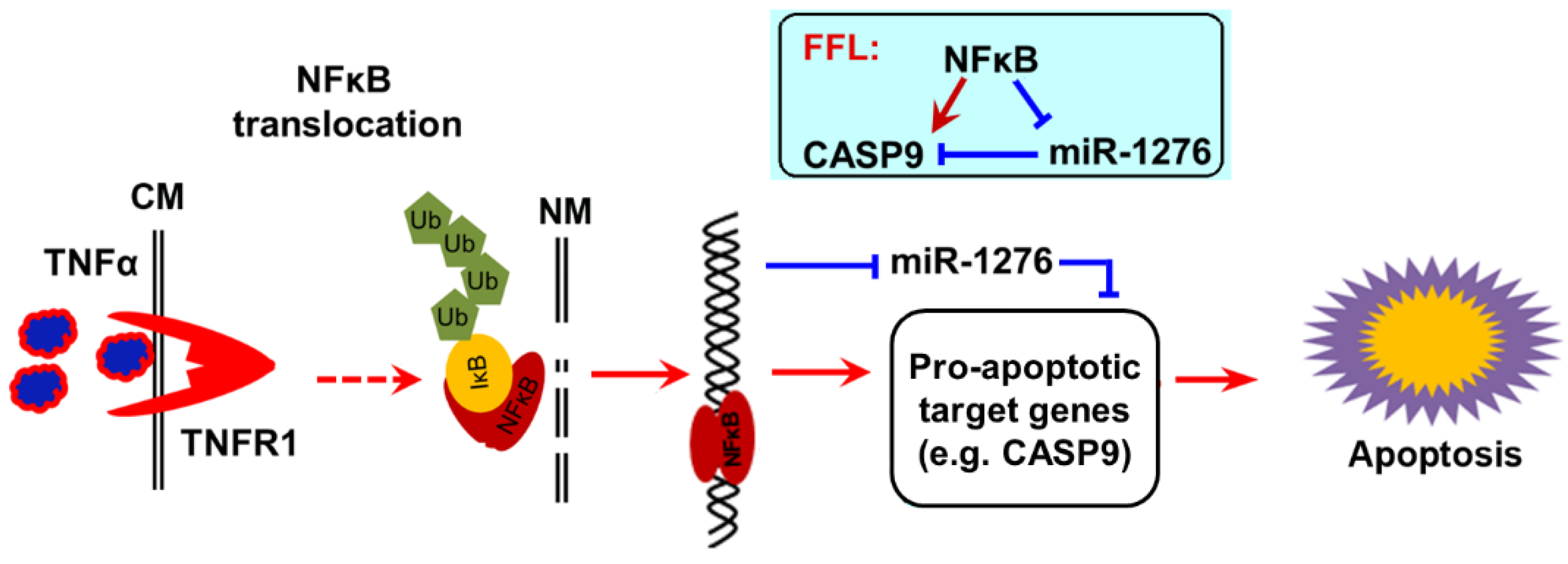
2.3. NF-κB Indirectly Regulates CASP9 Expression
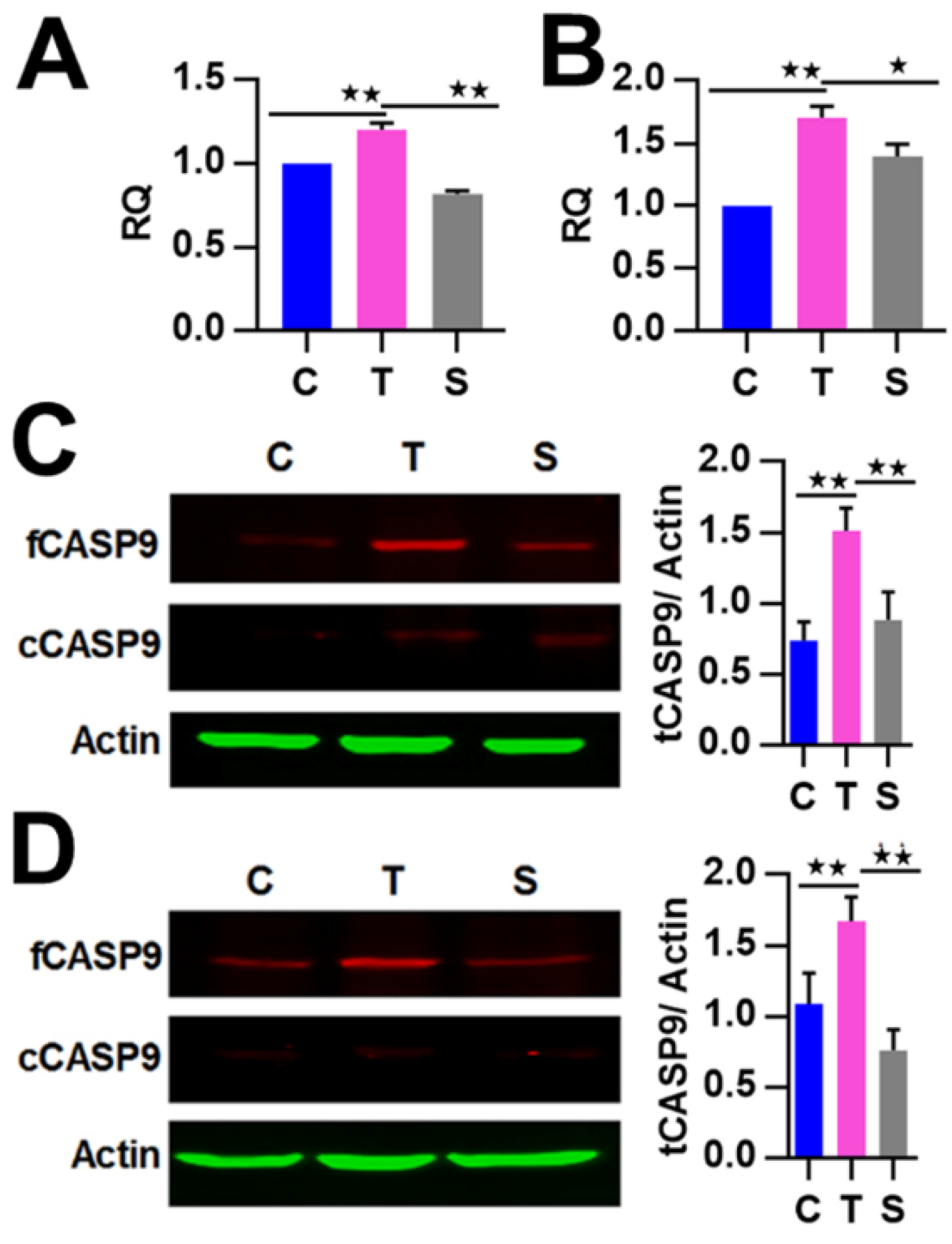
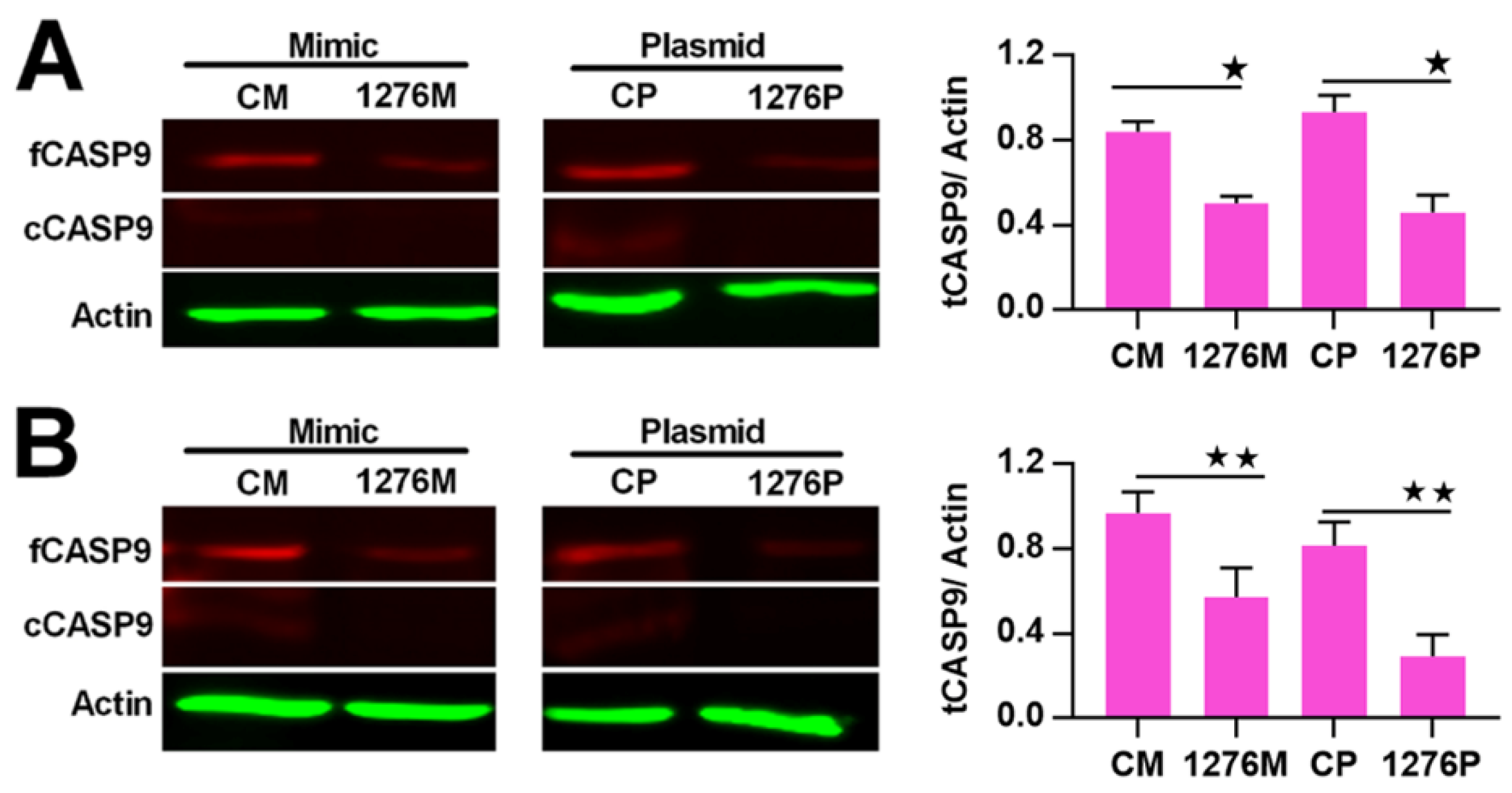
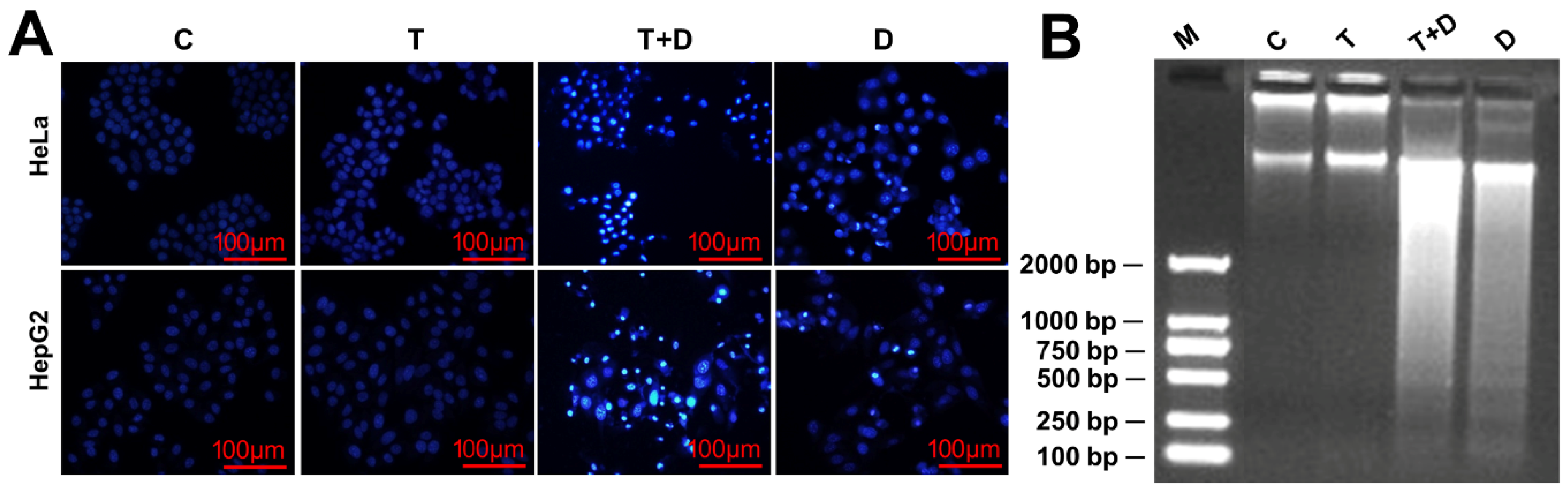
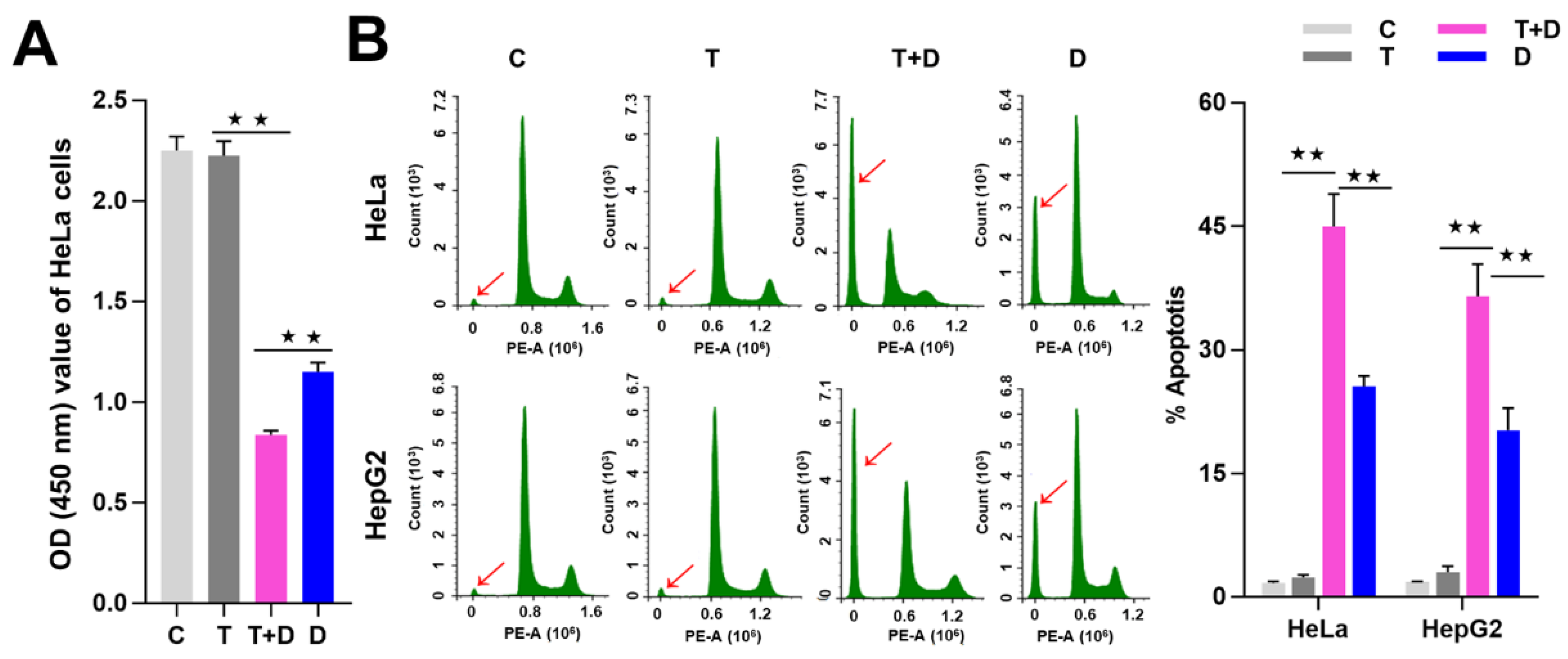
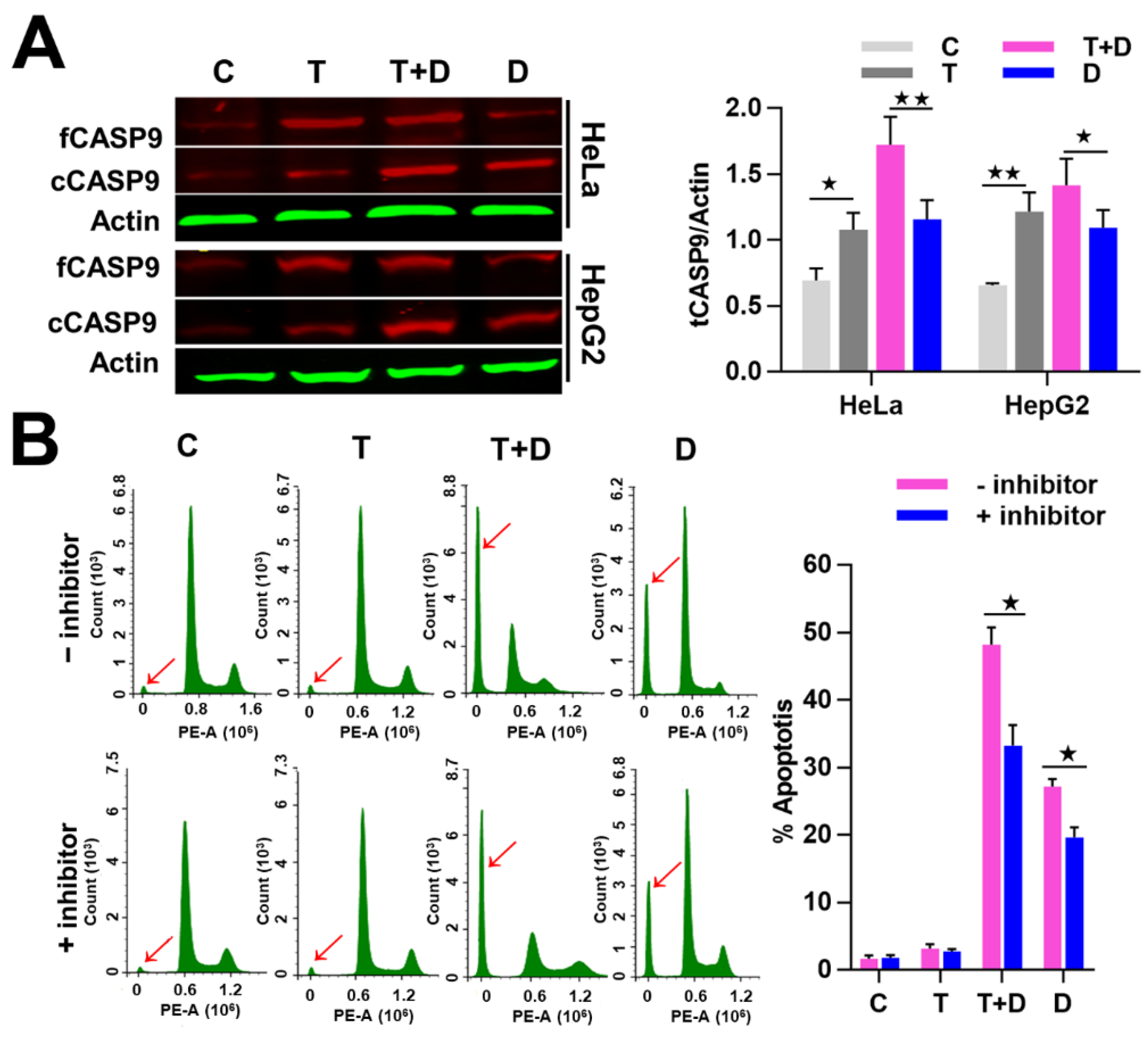
2.4. The Regulation of CASP9 by NF-κB-mediated FFL Contributes to the TNFα Promotion of DOX-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. ChIP-Seq Data Analysis
4.2. Cell Culture and Treatment
4.3. ChIP Expriment
4.4. ChIP-qPCR
4.5. Luciferase Reporter Assay
4.6. Cell Viability and Apoptosis Detection
4.7. Real-time Reverse Transcription-PCR
4.8. Western Blotassay
4.9. miRNA Target Prediction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BSs | NF-κB-binding sites |
| CASP | Caspase |
| cCASP9 | cleaved CASP9 |
| DOX | doxorubicin |
| fCASP9 | full-length CASP9 protein |
| TF | transcription factor |
| FFL | feed-forward loop |
| RQ | relative quantification |
| tCASP9 | total CASP9 |
| ChIP-Seq | Chromatin Immunoprecipitation-Sequencing |
References
- Zhou, F.; Xu, X.; Wu, J.; Wang, D.; Wang, J. NF-kappaB controls four genes encoding core enzymes of tricarboxylic acid cycle. Gene 2017, 621, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V.; Correa, R.G.; Ikawa, M.; Verma, I.M. Distinct roles of IkappaB proteins in regulating constitutive NF-kappaB activity. Nat. Cell Biol. 2005, 7, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Alabasi, G.; Sandaltzopoulos, R.; Marcu, K.B.; Kolettas, E. Roles of NF-kappaB Signaling in the Regulation of miRNAs Impacting on Inflammation in Cancer. Biomedicines 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 2012, 318, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Festjens, N.; Declercq, W.; Berghe, T.V.; Vandenabeele, P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007, 14, 44–55. [Google Scholar] [CrossRef]
- Yang, H.-J.; Wang, M.; Wang, L.; Cheng, B.-F.; Lin, X.-Y.; Feng, Z.-W. NF-κB Regulates Caspase-4 expression and sensitizes neuroblastoma cells to fas-induced apoptosis. PLoS ONE 2015, 10, e0117953. [Google Scholar] [CrossRef]
- Schauvliege, R.; Vanrobaeys, J.; Schotte, P.; Beyaert, R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 2002, 277, 41624–41630. [Google Scholar] [CrossRef]
- Kimura, M.; Haisa, M.; Uetsuka, H.; Takaoka, M.; Ohkawa, T.; Kawashima, R.; Yamatsuji, T.; Gunduz, M.; Kaneda, Y.; Tanaka, N.; et al. TNF combined with IFN-alpha accelerates NF-kappaB-mediated apoptosis through enhancement of Fas expression in colon cancer cells. Cell Death Differ. 2003, 10, 718–728. [Google Scholar] [CrossRef]
- Jennewein, C.; Karl, S.; Baumann, B.; Micheau, O.; Debatin, K.; Fulda, S. Identification of a novel pro-apoptotic role of NF-κB in the regulation of TRAIL-and CD95-mediated apoptosis of glioblastoma cells. Oncogene 2012, 31, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Ernst, M.K.; Rice, N.R.; Vousden, K.H. Role of NF-kappaB in p53-mediated programmed cell death. Nature 2000, 404, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, J.Y.; Wu, G.S. Tumor necrosis factor-related apoptosis-inducing ligand is required for tumor necrosis factor alpha-mediated sensitization of human breast cancer cells to chemotherapy. Cancer Res. 2006, 66, 10092–10099. [Google Scholar] [CrossRef] [PubMed]
- Karl, S.; Pritschow, Y.; Volcic, M.; Häcker, S.; Baumann, B.; Wiesmüller, L.; Debatin, K.M.; Fulda, S. Identification of a novel pro-apopotic function of NF-κB in the DNA damage response. J. Cell. Mol. Med. 2009, 13, 4239–4256. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, J.; An, Y.; Yang, Y.; Zhang, S.Q. Doxorubicin-induced apoptosis in H9c2 cardiomyocytes by NF-kappaB dependent PUMA upregulation. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2323–2329. [Google Scholar] [PubMed]
- Xing, Y.; Yang, Y.; Zhou, F.; Wang, J. Characterization of genome-wide binding of NF-kappaB in TNFalpha-stimulated HeLa cells. Gene 2013, 526, 142–149. [Google Scholar] [CrossRef]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.J.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.H.; Kung, A.L.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef]
- Zhao, B.; Barrera, L.A.; Ersing, I.; Willox, B.; Schmidt, S.C.; Greenfeld, H.; Zhou, H.; Mollo, S.B.; Shi, T.T.; Takasaki, K.; et al. The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014, 8, 1595–1606. [Google Scholar] [CrossRef]
- Kasowski, M.; Grubert, F.; Heffelfinger, C.; Hariharan, M.; Asabere, A.; Waszak, S.M.; Habegger, L.; Rozowsky, J.; Shi, M.; Urban, A.E.; et al. Variation in transcription factor binding among humans. Science 2010, 328, 232–235. [Google Scholar] [CrossRef]
- Wong, D.; Teixeira, A.; Oikonomopoulos, S.; Humburg, P.; Lone, I.N.; Saliba, D.; Siggers, T.; Bulyk, M.; Angelov, D.; Dimitrov, S. Extensive characterization of NF-κB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011, 12, R70. [Google Scholar] [CrossRef]
- Bally, A.P.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-κB Regulates PD-1 Expression in Macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xu, X.; Wang, D.; Wu, J.; Wang, J. Identification of novel NF-kappaB transcriptional targets in TNFalpha-treated HeLa and HepG2 cells. Cell Biol. Int. 2017, 41, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wang, W.; Xing, Y.; Wang, T.; Xu, X.; Wang, J. NF-kappaB target microRNAs and their target genes in TNFalpha-stimulated HeLa cells. Biochim. Biophys. Acta 2014, 1839, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, K.; Wieder, T.; Tamm, I.; Müller, A.; Essmann, F.; Geilen, C.-C.; Schulze-Osthoff, K.; Dörken, B.; Daniel, P.-T. Tumor necrosis factor α sensitizes malignant cells to chemotherapeutic drugs via the mitochondrial apoptosis pathway independently of caspase-8 and NF-κB. Oncogene 2004, 23, 6743–6759. [Google Scholar] [CrossRef][Green Version]
- Wu, H.; Lozano, G. NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J. Biol. Chem. 1994, 269, 20067–20074. [Google Scholar]
- Wang, P.; Qiu, W.; Dudgeon, C.; Liu, H.; Huang, C.; Zambetti, G.P.; Yu, J.; Zhang, L. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death Differ. 2009, 16, 1192–1202. [Google Scholar] [CrossRef]
- Cao, W.; Chi, W.H.; Wang, J.; Tang, J.J.; Lu, Y.J. TNF-alpha promotes Doxorubicin-induced cell apoptosis and anti-cancer effect through downregulation of p21 in p53-deficient tumor cells. Biochem. Biophys. Res. Commun. 2005, 330, 1034–1040. [Google Scholar] [CrossRef]
- Shinoura, N.; Sakurai, S.; Asai, A.; Kirino, T.; Hamada, H. Transduction of Apaf-1 or caspase-9 induces apoptosis in A-172 cells that are resistant to p53-mediated apoptosis. Biochem. Biophys. Res. Commun. 2000, 272, 667–673. [Google Scholar] [CrossRef]
- Corvaro, M.; Fuoco, C.; Wagner, M.; Cecconi, F. Analysis of apoptosome dysregulation in pancreatic cancer and of its role in chemoresistance. Cancer Biol. Ther. 2007, 6, 209–217. [Google Scholar] [CrossRef][Green Version]
- Druškovič, M.; Šuput, D.; Milisav, I. Overexpression of caspase-9 triggers its activation and apoptosis in vitro. Croat. Med. J. 2006, 47, 832–840. [Google Scholar]
- Chen, F.-H.; Lu, N.; Zhang, H.-W.; Zhao, L.; He, L.-C.; Sun, H.-P.; You, Q.-D.; Li, Z.-Y.; Guo, Q.-L. LYG-202 augments tumor necrosis Factor-α-Induced apoptosis via attenuating casein kinase 2-Dependent nuclear Factor-κB pathway in HepG2 cells. Mol. Pharmacol. 2012, 82, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Higashitsuji, H.; Nagao, T.; Nonoguchi, K.; Fujii, S.; Itoh, K.; Fujita, J. A novel protein overexpressed in hepatoma accelerates export of NF-kappa B from the nucleus and inhibits p53-dependent apoptosis. Cancer Cell 2002, 2, 335–346. [Google Scholar] [CrossRef]
- Baskerville, S.; Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, S.T.; Fuh, C.S.; Juan, H.F.; Huang, H.C. Coregulation of transcription factors and microRNAs in human transcriptional regulatory network. BMC Bioinform. 2011, 12, S41. [Google Scholar] [CrossRef]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and local architecture of the mammalian microRNA–transcription factor regulatory network. PLoS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef]
- Lim, C.A.; Yao, F.; Wong, J.J.; George, J.; Xu, H.; Chiu, K.P.; Sung, W.K.; Lipovich, L.; Vega, V.B.; Chen, J.; et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol. Cell 2007, 27, 622–635. [Google Scholar] [CrossRef]
- Zhou, F.; Xing, Y.; Xu, X.; Yang, Y.; Zhang, J.; Ma, Z.; Wang, J. NBPF is a potential DNA-binding transcription factor that is directly regulated by NF-kappaB. Int. J. Biochem. Cell Biol. 2013, 45, 2479–2490. [Google Scholar] [CrossRef]
- Xie, J.J.; Zhou, F.; Li, E.M.; Jiang, H.; Du, Z.P.; Lin, R.; Fang, D.S.; Xu, L.Y. FW523-3, a novel lipopeptide compound, induces apoptosis in cancer cells. Mol. Med. Rep. 2011, 4, 759–763. [Google Scholar] [PubMed]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Grosse, I.; Hatzigeorgiou, A.G. Functional microRNA targets in protein coding sequences. Bioinformatics 2012, 28, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk—Database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BSs | FE | κB Number | Location |
|---|---|---|---|
| BS1 | 20.579 | 44 | 3′ region |
| BS2 | 21.720 | 23 | 3′ region |
| BS3 | 21.236 | 55 | 3′ region |
| BS4 | 34.436 | 14 | 3′ region |
| BS5 | 30.491 | 8 | 3′ region |
| BS6 | 21.982 | 8 | 3′ region |
| BS7 | 22.187 | 21 | 3′ region |
| BS8 | 21.805 | 3 | 3′ region |
| BS9 | 20.638 | 31 | 3′ region |
| BS10 | 27.507 | 17 | 5′ region |
| BS11 | 21.918 | 19 | 5′ region |
| Average | 24.045 | 22.091 |
| BSs | FE | κB Number | Location |
|---|---|---|---|
| BS1 | 22.87 | 3 | 3′ region |
| BS2 | 21.869 | 4 | 3′ region |
| BS3 | 20.815 | 10 | 3′ region |
| BS4 | 32.358 | 28 | 3′ region |
| BS5 | 20.386 | 68 | 3′ region |
| BS6 | 21.175 | 77 | Internal |
| BS7 | 25.373 | 55 | Internal |
| BS8 | 23.653 | 30 | Internal |
| BS9 | 28.99 | 35 | 5′ region |
| BS10 | 25.738 | 4 | 5′ region |
| BS11 | 29.767 | 2 | 5′ region |
| Average | 24.818 | 28.727 |
| miRWalk | TargetScan | DIANA-microT-CDS | |
|---|---|---|---|
| Binding type | 7 mer | 7 mer-1A/7 mer-m8 | 7 mer/6 mer/6 mer/7 mer/6 mer/6 mer/7 mer(CDS) |
| Potential binding site | 1 | 1 | 7 (6 in 3′ UTR +1 in CDS) |
| Region | 3′ UTR | 3′ UTR | 3′ UTR and CDS |
| Confidence | P value = 0.041 | Context + +score = −0.11 | miTG core = 0.766 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, F.; Li, Y.; Huang, Y.; Wu, J.; Wu, Q.; Zhu, H.; Wang, J. Upregulation of CASP9 through NF-κB and Its Target MiR-1276 Contributed to TNFα-Promoted Apoptosis of Cancer Cells Induced by Doxorubicin. Int. J. Mol. Sci. 2020, 21, 2290. https://doi.org/10.3390/ijms21072290
Zhou F, Li Y, Huang Y, Wu J, Wu Q, Zhu H, Wang J. Upregulation of CASP9 through NF-κB and Its Target MiR-1276 Contributed to TNFα-Promoted Apoptosis of Cancer Cells Induced by Doxorubicin. International Journal of Molecular Sciences. 2020; 21(7):2290. https://doi.org/10.3390/ijms21072290
Chicago/Turabian StyleZhou, Fei, Yun Li, Yisheng Huang, Jian Wu, Qinhan Wu, Hui Zhu, and Jinke Wang. 2020. "Upregulation of CASP9 through NF-κB and Its Target MiR-1276 Contributed to TNFα-Promoted Apoptosis of Cancer Cells Induced by Doxorubicin" International Journal of Molecular Sciences 21, no. 7: 2290. https://doi.org/10.3390/ijms21072290
APA StyleZhou, F., Li, Y., Huang, Y., Wu, J., Wu, Q., Zhu, H., & Wang, J. (2020). Upregulation of CASP9 through NF-κB and Its Target MiR-1276 Contributed to TNFα-Promoted Apoptosis of Cancer Cells Induced by Doxorubicin. International Journal of Molecular Sciences, 21(7), 2290. https://doi.org/10.3390/ijms21072290