Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel

 , ,
, ,  ,
, 

Abstract
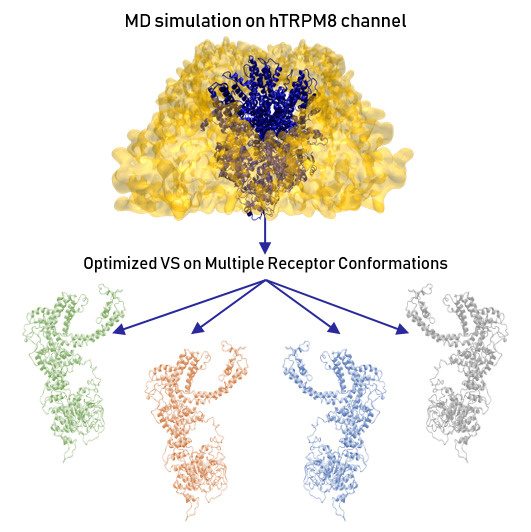
1. Introduction
2. Results
2.1. hTRPM8 Homology Model
2.2. MD Simulation
2.3. Frame Selection
2.4. Virtual Screening Campaigns
2.4.1. Virtual Screening Results for Each Monomer of Each Frame
2.4.2. Virtual Screening Results Combining the Four Monomers of a Single Frame
2.4.3. Virtual Screening Results Combining the Four Monomers of all Selected Frames
3. Discussion
4. Materials and Methods
4.1. Monomer Generation
4.2. Tetramer Assembly and MD Simulation
4.3. Virtual Screening Analyses
4.4. Hardware Details
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TRPM8 | Transient Receptor Potential Cation Channel Subfamily M (Melastatin) 8 |
| PIP2 | Phosphatidylinositol 4,5-Bisphosphate |
| MD | Molecular Dynamics |
| VS | Virtual Screening |
| EFO | Enrichment Factor Optimization |
| RMSD | Root-Mean-Square Deviation |
| EF | Enrichment Factor |
| MLPInS MRC | Molecular Lipophilicity Potential Interaction Score Multiple Receptor Conformations |
References
- Voet, T.; Owsianik, G.; Nilius, B. TRPM8. Handb. Exp Pharm. 2007, 179, 329–344. [Google Scholar]
- Almaraz, L.; Manenschijn, J.-A.; de la Peña, E.; Viana, F. TRPM8. Mammalian Transient Receptor Potential (TRP) cation Channels; Nilius, B., Flockerzi, V., Eds.; Springer: Berlin, Germany, 2014. [Google Scholar]
- García-Ávila, M.; Islas, L.D. What is new about mild temperature sensing? A review of recent findings. Temperature 2019, 6, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Señarís, R.; Ordás, P.; Reimúndez, A.; Viana, F. Mammalian cold TRP channels: Impact on thermoregulation and energy homeostasis. Pflug. Arch. 2018, 470, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Gupta, R.; Jordt, S.E.; Chen, Y.; Liedtke, W.B. Regulation of Pain and Itch by TRP Channels. Neurosci. Bull. 2018, 34, 120–142. [Google Scholar] [CrossRef]
- Pérez de Vega, M.J.; Gómez-Monterrey, I.; Ferrer-Montiel, A.; González-Muñiz, R. Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J. Med. Chem. 2016, 59, 10006–10029. [Google Scholar] [CrossRef]
- Benemei, S.; Dussor, G. TRP Channels and Migraine: Recent Developments and New Therapeutic Opportunities. Pharmaceuticals 2019, 12, 54. [Google Scholar] [CrossRef]
- Yang, J.M.; Wei, E.T.; Kim, S.J.; Yoon, K.C. TRPM8 Channels and Dry Eye. Pharmaceuticals 2018, 11, 125. [Google Scholar] [CrossRef]
- Beckers, A.B.; Weerts, Z.Z.R.M.; Helyes, Z.; Masclee, A.A.M.; Keszthelyi, D. Review article: Transient receptor potential channels as possible therapeutic targets in irritable bowel syndrome. Aliment. Pharm. Ther. 2017, 46, 938–952. [Google Scholar] [CrossRef]
- Bonvini, S.J.; Belvisi, M.G. Cough and airway disease: The role of ion channels. Pulm. Pharm. Ther. 2017, 47, 21–28. [Google Scholar] [CrossRef]
- Rohacs, T.; Nilius, B. Regulation of transient receptor potential (TRP) channels by phosphoinositides. Pflug. Arch. 2007, 455, 157–168. [Google Scholar] [CrossRef]
- Moran, M.M. TRP Channels as Potential Drug Targets. Annu. Rev. Pharm. Toxicol. 2018, 58, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Bandell, M.; Dubin, A.E.; Petrus, M.J.; Orth, A.; Mathur, J.; Hwang, S.W.; Patapoutian, A. High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nat. Neurosci. 2006, 9, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rosasco, M.G.; Gordon, S.E. TRP Channels: What Do They Look Like. In Neurobiology of TRP Channels, 2nd ed.; Emir, T.L.R., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017; Chapter 1. [Google Scholar]
- González-Muñiz, R.; Bonache, M.A.; Martín-Escura, C.; Gómez-Monterrey, I. Recent Progress in TRPM8 Modulation: An Update. Int. J. Mol. Sci. 2019, 20, 2618. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Marconi, C.; Bettinelli, I.; Vistoli, G. Comparative modeling of the quaternary structure for the human TRPM8 channel and analysis of its binding features. Biochim. Biophys. Acta 2009, 1788, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Beccari, A.R.; Gemei, M.; Lo Monte, M.; Menegatti, N.; Fanton, M.; Pedretti, A.; Bovolenta, S.; Nucci, C.; Molteni, A.; Rossignoli, A.; et al. Novel selective, potent naphthyl TRPM8 antagonists identified through a combined ligand- and structure-based virtual screening approach. Sci. Rep. 2017, 7, 10999. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, 6430. [Google Scholar] [CrossRef]
- Bertamino, A.; Iraci, N.; Ostacolo, C.; Ambrosino, P.; Musella, S.; Di Sarno, V.; Ciaglia, T.; Pepe, G.; Sala, M.; Soldovieri, M.V.; et al. Identification of a Potent Tryptophan-Based TRPM8 Antagonist With in Vivo Analgesic Activity. J. Med. Chem. 2018, 61, 6140–6152. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Beccari, A.R.; Cavazzoni, C.; Beato, C.; Costantino, G. LiGen: A high performance workflow for chemistry driven de novo design. J. Chem. Inf. Model. 2013, 53, 1518–1527. [Google Scholar] [CrossRef]
- Pedretti, A.; Granito, C.; Mazzolari, A.; Vistoli, G. Structural Effects of Some Relevant Missense Mutations on the MECP2-DNA Binding: A MD Study Analyzed by Rescore+, a Versatile Rescoring Tool of the VEGA ZZ Program. Mol. Inform. 2016, 35, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Mazzolari, A.; Vistoli, G.; Testa, B.; Pedretti, A. Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program. Molecules 2018, 23, 2955. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Niu, Y.; Yao, X.; Ji, H. Importance of protein flexibility in ranking ERK2 Type I1/2 inhibitor affinities: A computational study. RSC Adv. 2019, 9, 12441–12454. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Vistoli, G. Rescoring and Linearly Combining: A Highly Effective Consensus Strategy for Virtual Screening Campaigns. Int. J. Mol. Sci. 2019, 20, 2060. [Google Scholar] [CrossRef]
- Vistoli, G.; Mazzolari, A.; Testa, B.; Pedretti, A. Binding Space Concept: A New Approach To Enhance the Reliability of Docking Scores and Its Application to Predicting Butyrylcholinesterase Hydrolytic Activity. J. Chem. Inf. Model. 2017, 57, 1691–1702. [Google Scholar] [PubMed]
- Vistoli, G.; Pedretti, A.; Mazzolari, A.; Testa, B. In silico prediction of human carboxylesterase-1 (hCES1) metabolism combining docking analyses and MD simulations. Bioorg. Med. Chem. 2010, 18, 320–329. [Google Scholar] [CrossRef]
- Evangelista, W.; Ellingson, S.R.; Smith, J.C.; Baudry, J.Y. Ensemble Docking in Drug Discovery: How Many Protein Configurations from Molecular Dynamics Simulations Are Needed to Reproduce Known Ligand Binding? J. Phys. Chem. B 2019, 123, 5189–5195. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—a program to check the stereochemical quality of protein structures. J. App. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA: A versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graph. Model. 2002, 21, 47–49. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular Recognition of Receptor Sites Using a Genetic Algorithm with a Description of Desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S1−S6 | TRP | N-TER | C-TER | Average | |
|---|---|---|---|---|---|
| Chain A | 2.56 | 1.60 | 3.65 | 2.55 | 2.59 |
| Chain B | 2.89 | 2.12 | 4.24 | 4.43 | 3.42 |
| Chain C | 2.91 | 2.20 | 4.25 | 3.36 | 3.18 |
| Chain D | 3.39 | 2.38 | 3.93 | 3.44 | 3.28 |
| Binding Site | 6NR2 | 6NR3 | |||
|---|---|---|---|---|---|
| Frame | RMSD | Frame | RMSD | ||
| Chain A | Backbone | 566 | 3.27 | 999 | 2.71 |
| Side chains | 1047 | 3.81 | 1000 | 3.23 | |
| Chain B | Backbone | 990 | 2.70 | 991 | 2.77 |
| Side chains | 1049 | 3.72 | 1049 | 3.74 | |
| Chain C | Backbone | 562 | 2.09 | 715 | 2.25 |
| Side chains | 562 | 3.20 | 655 | 3.39 | |
| Chain D | Backbone | 553 | 2.58 | 1246 | 2.27 |
| Side chains | 900 | 3.22 | 1244 | 3.29 | |
| Table 3A | Frame | PLANTS ChemPLP | GOLD ChemPLP | GOLD GoldScore | GOLD ASP | LiGen™ | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 6NR2 WS-12 | ||||||||||
| Chain A | 566 | 7.62 | 7.96 | 8.18 | 7.83 | 3.55 | ||||
| 1047 | 8.07 | 9.07 | 8.24 | 8.58 | 3.35 | |||||
| Chain B | 990 | 9.63 | 7.51 | 7.50 | 5.12 | 3.51 | ||||
| 1049 | 8.30 | 7.71 | 7.17 | 7.96 | 3.31 | |||||
| Chain C | 562 | 7.88 | 3.54 | 3.59 | 4.42 | 3.79 | ||||
| Chain D | 553 | 11.25 | 8.45 | 9.09 | 8.41 | 3.75 | ||||
| 900 | 11.86 | 11.30 | 11.46 | 12.01 | nn | |||||
| 6NR3 Icilin | ||||||||||
| Chain A | 999 | 6.51 | 7.04 | 7.84 | 7.28 | 6.85 | ||||
| 1000 | 7.20 | 6.93 | 8.20 | 7.07 | 7.08 | |||||
| Chain B | 991 | 8.20 | 6.54 | 7.96 | 8.93 | 3.30 | ||||
| 1049 | 9.10 | 3.71 | 8.33 | 3.18 | 3.06 | |||||
| Chain C | 715 | 12.31 | 12.37 | 5.12 | 5.12 | 7.34 | ||||
| 655 | 4.78 | 8.08 | 8.33 | 6.01 | 4.77 | |||||
| Chain D | 1246 | 14.06 | 7.64 | 4.28 | 7.91 | nn | ||||
| 1244 | 14.75 | 4.78 | 4.77 | 5.06 | nn | |||||
| Table 3B | Frame | PLANTS ChemPLP | GOLD ChemPLP | GOLD GoldScore | GOLD ASP | LiGen™ | Glide | |||
| 6NR2 WS-12 | ||||||||||
| Chain A | 566 | 7.99 | 4.92 | 5.25 | 7.80 | 3.82 | 4.87 | |||
| 1047 | 5.73 | 9.00 | 6.03 | 8.07 | 7.61 | 5.58 | ||||
| Chain B | 990 | 8.99 | 2.69 | 2.90 | 9.90 | 7.11 | 6.26 | |||
| 1049 | 8.05 | 8.42 | 6.04 | 8.30 | 2.88 | 7.87 | ||||
| Chain C | 562 | 7.73 | 7.53 | 7.74 | 4.31 | 7.69 | 8.95 | |||
| 562 | 7.73 | 7.53 | 7.74 | 4.31 | 7.69 | 8.95 | ||||
| Chain D | 553 | 11.27 | 8.20 | 10.83 | 8.52 | 3.56 | 6.13 | |||
| 900 | 12.41 | 8.52 | 8.38 | 8.42 | nn | nn | ||||
| 6NR3 Icilin | ||||||||||
| Chain A | 999 | 8.00 | 7.54 | 8.08 | 7.28 | 6.90 | 7.35 | |||
| 1000 | 6.68 | 8.86 | 8.19 | 7.89 | 6.95 | 5.63 | ||||
| Chain B | 991 | 5.30 | 7.65 | 7.34 | 8.07 | 7.61 | 7.38 | |||
| 1049 | 5.95 | 7.75 | 7.90 | 7.68 | 3.11 | 6.74 | ||||
| Chain C | 715 | 11.37 | 5.42 | 5.03 | 11.86 | 2.25 | 8.44 | |||
| 655 | 8.22 | 8.99 | 8.46 | 8.04 | 4.70 | 8.52 | ||||
| Chain D | 1246 | 13.89 | 4.97 | 4.59 | 5.13 | nn | nn | |||
| 1244 | 11.89 | 4.98 | 6.72 | 5.28 | nn | nn | ||||
| Monomer | LiGen™ 1 score | LiGen™ EFO | Gold 1 score | Gold EFO | Plants 1 score | Plants EFO | Mean 1 score | Mean EFO |
|---|---|---|---|---|---|---|---|---|
| 562 (overall mean = 10.23) | ||||||||
| Chain A | 5.76 | 34.54 | 0.00 | 9.59 | 5.76 | 30.71 | 3.84 | 24.95 |
| Chain B | 3.84 | 26.87 | 0.00 | 1.92 | 0.00 | 5.76 | 1.28 | 11.52 |
| Chain C | 11.51 | 28.79 | 0.00 | 1.92 | 1.92 | 13.43 | 4.48 | 14.71 |
| Chain D | 3.84 | 40.30 | 1.92 | 5.75 | 0.00 | 11.51 | 1.92 | 19.19 |
| Mean | 6.24 | 32.62 | 0.48 | 4.80 | 1.92 | 15.35 | 2.88 | 17.59 |
| 990 (overall mean = 9.16) | ||||||||
| Chain A | 15.35 | 65.25 | 13.42 | 13.42 | 5.76 | 11.54 | 11.51 | 30.07 |
| Chain B | 9.60 | 17.27 | 0.00 | 1.92 | 0.00 | 1.92 | 3.20 | 7.04 |
| Chain C | 3.84 | 17.27 | 0.00 | 1.00 | 1.92 | 9.60 | 1.92 | 9.29 |
| Chain D | 3.84 | 21.11 | 0.00 | 3.84 | 0.00 | 1.92 | 1.28 | 8.96 |
| Mean | 8.16 | 30.23 | 3.36 | 5.05 | 1.92 | 6.25 | 4.48 | 13.84 |
| 1049 (overall mean = 18.43) | ||||||||
| Chain A | 26.87 | 57.57 | 1.92 | 1.92 | 7.68 | 38.38 | 12.16 | 32.62 |
| Chain B | 5.76 | 40.30 | 0.00 | 3.84 | 1.92 | 19.19 | 2.56 | 21.11 |
| Chain C | 13.43 | 28.79 | 9.62 | 9.59 | 3.84 | 26.87 | 8.96 | 21.75 |
| Chain D | 53.73 | 55.65 | 0.00 | 1.00 | 1.92 | 32.62 | 18.55 | 29.76 |
| Mean | 24.95 | 45.58 | 2.89 | 4.09 | 3.84 | 29.27 | 10.56 | 26.31 |
| Global Mean | 13.11 | 36.14 | 2.24 | 4.64 | 2.56 | 16.95 | 5.97 | 19.25 |
| Equation | Frame | Docking Tool | Model | EF 1% |
|---|---|---|---|---|
| 1 | 562 | LiGen™ | 1.00 PS_D – 2.39 MLPINS_A + 19.76 PLP_A – 13.08 MLPINS_C | 46.15 |
| 2 | 562 | PLANTS | -1.00 MLPINS_A – 3.02 PLP95NORM_HEVATMS_C | 34.62 |
| 3 | 990 | LiGen™ | -1.00 MLPINS_B + 0.13 CHEMPLP_B + 54.59 ContactsNORM_HEVATMS_A – 5.39 Xscore_A | 59.23 |
| 4 | 990 | PLANTS | -1.00 MLPINS_B + 534.45 ContactsNORM_HEVATMS_A – 14.77 PLP95NORM_HEVATMS_A | 32.69 |
| 5 | 1049 | LiGen™ | 1.00 Contacts_A – 14.20 PLP95NORM_HEVATMS_A – 13.66 MLPINS_C – 5.35 Csopt_D | 57.69 |
| 6 | 1049 | PLANTS | 1.00 ContactsNORM_WEIGHT_C – 0.0092 MLPINS_D + 0.041 MLPINS_D – 0.10 PLP95NORM_HEVATMS_A | 48.08 |
| 7 | All | PLANTS (1) | MLPINS_A_562 | 23.08 |
| 8 | All | PLANTS (2) | -1.00 MLPINS_A_562 – 4.59 XscoreHM_D_1049 | 38.38 |
| 9 | All | PLANTS (3) | -1.00 MLPINS_A_562 + 5.34 MLPINS_C_1049 + 50.34 ContactsNORM_HEVATMS_D_1049 | 49.89 |
| 10 | All | PLANTS (4) | -1.00 MLPINS_A_562 + 33.10 ContactsNORM_HEVATMS_D_562 + 4,33 MLPINS_C_1049 – 0,12 MLPINS_C_1049 | 51.82 |
| 11 | All | LiGen™ (1) | ContactsNORM_HEVATMS_A_990 | 38.38 |
| 12 | All | LiGen™ (2) | 1.00 PS_D_562 + 0.46 ContactsNORM_HEVATMS_A_990 | 55.65 |
| 13 | All | LiGen™ (3) | -100 MLPINS_B_990 + 0.84 Contacts_A_990 – 1.84 CS_D_1049 | 65.28 |
| 14 | All | LiGen™ (4) | 1.00 ContactsNORM_WEIGHT_D_990 – 0.042 MLPINS_B_990 + 0.053 Contacts_A_990 – 0.031 Csopt_D_1049 | 67.11 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talarico, C.; Gervasoni, S.; Manelfi, C.; Pedretti, A.; Vistoli, G.; Beccari, A.R. Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel. Int. J. Mol. Sci. 2020, 21, 2265. https://doi.org/10.3390/ijms21072265
Talarico C, Gervasoni S, Manelfi C, Pedretti A, Vistoli G, Beccari AR. Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel. International Journal of Molecular Sciences. 2020; 21(7):2265. https://doi.org/10.3390/ijms21072265
Chicago/Turabian StyleTalarico, Carmine, Silvia Gervasoni, Candida Manelfi, Alessandro Pedretti, Giulio Vistoli, and Andrea R. Beccari. 2020. "Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel" International Journal of Molecular Sciences 21, no. 7: 2265. https://doi.org/10.3390/ijms21072265
APA StyleTalarico, C., Gervasoni, S., Manelfi, C., Pedretti, A., Vistoli, G., & Beccari, A. R. (2020). Combining Molecular Dynamics and Docking Simulations to Develop Targeted Protocols for Performing Optimized Virtual Screening Campaigns on the hTRPM8 Channel. International Journal of Molecular Sciences, 21(7), 2265. https://doi.org/10.3390/ijms21072265