Iron Metabolism in Cancer Progression



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Historical Bases
2.1. Relationship Between Iron Metabolism and Cancer
2.2. Hereditary Hemochromatosis, a Genetic Iron-Overload Disorder, and Its Association with Cancer
2.3. Mini-Summary
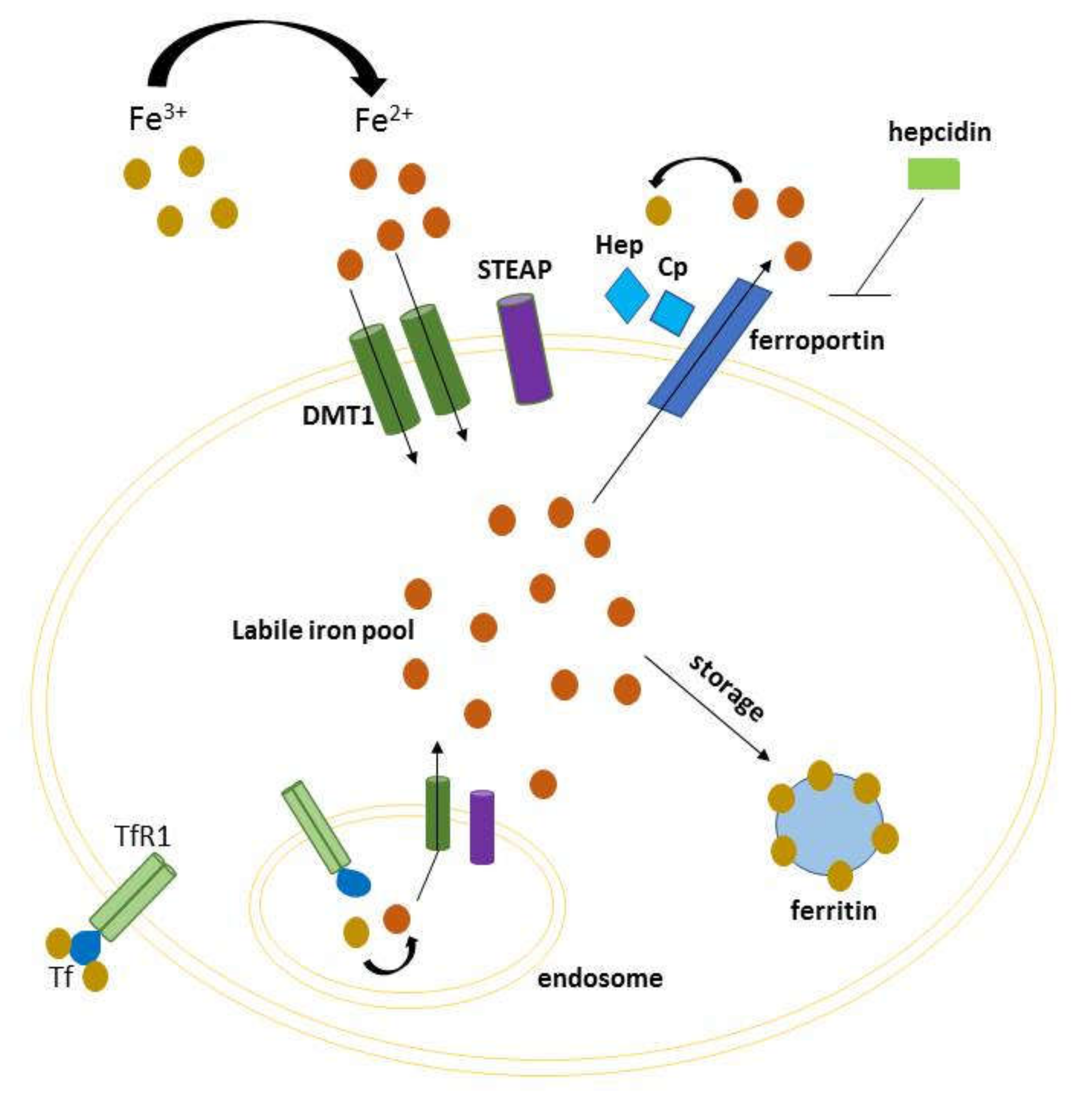
3. Molecular Mechanisms
3.1. The Role of Iron in Oxidative Stress
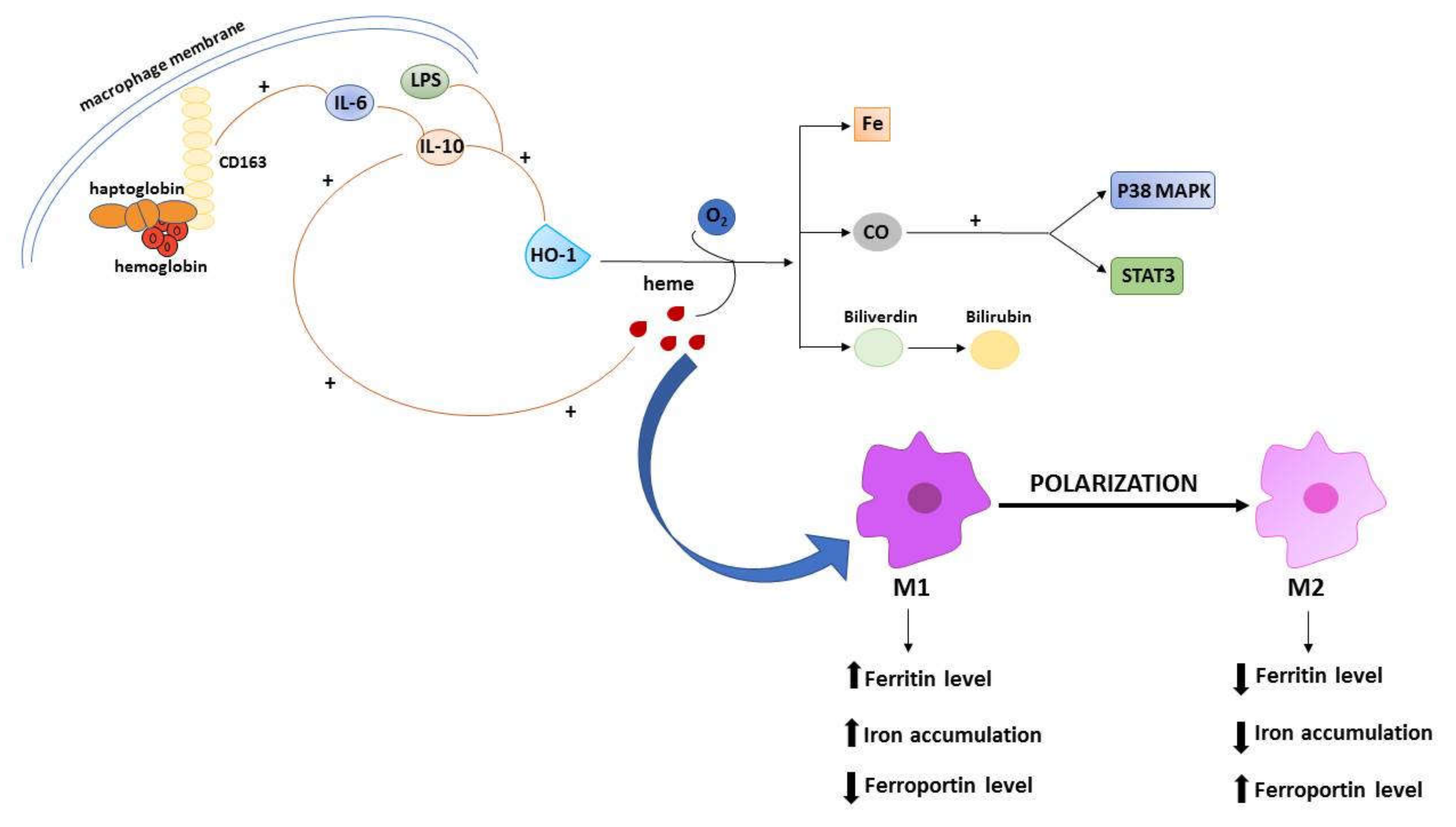
3.2. Hemeoxygenase-1 Induction in Cancer Progression
3.3. The Role of Tumor-Associated Macrophages in Iron Metabolism
3.4. Mini-Summary
4. Interplay Between Iron Metabolism and Immune System in Tumor
Mini-Summary
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Milto, I.V.; Suhodolo, I.V.; Prokopieva, V.D.; Klimenteva, T.K. Molecular and Cellular Bases of Iron Metabolism in Humans. Biochemistry 2016, 81, 549–564. [Google Scholar] [CrossRef]
- Imam, M.U.; Zhang, S.; Ma, J.; Wang, H.; Wang, F. Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress. Nutrients 2017, 9, 671. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, B.K.; Lu, Y.; Frazer, D.M.; Darshan, D.; Wilkins, S.J.; Dunn, L.; Loguinov, A.V.; Kogan, S.C.; Matak, P.; Chen, H.; et al. Severe Iron Metabolism Defects in Mice With Double Knockout of the Multicopper Ferroxidases Hephaestin and Ceruloplasmin. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 405–427. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.; Wallace, D.F.; Subramaniam, V.N. Hepcidin: Regulation of the master iron regulator. Biosci. Rep. 2015, 35, 1–12. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. 1), S6–S20. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Mertens, C.; Tomat, E.; Brüne, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Ironing out cancer. Cancer Res. 2011, 71, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, F. Iron homeostasis and tumorigenesis: Molecular mechanisms and therapeutic opportunities. Protein Cell 2015, 6, 88–100. [Google Scholar] [CrossRef]
- Ludwig, H.; Evstatiev, R.; Kornek, G.; Aapro, M.; Bauernhofer, T.; Buxhofer-Ausch, V.; Fridrik, M.; Geissler, D.; Geissler, K.; Gisslinger, H.; et al. Iron metabolism and iron supplementation in cancer patiens. Wien. Klin. Wochenschr. 2015, 127, 907–919. [Google Scholar] [CrossRef]
- Bystrom, L.M.; Rivella, S. Cancer cells with irons in the fire. Free Radic. Biol. Med. 2015, 79, 337–342. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, B.; Zhang, L.; Wang, S.; Dong, D.; Lv, H.; Shang, P. Alterations in Cellular Iron Metabolism Provide More Therapeutic Opportunities for Cancer. Int. J. Mol. Sci. 2018, 19, 1545. [Google Scholar] [CrossRef]
- Adachi, M.; Kai, K.; Yamaji, K.; Ide, T.; Noshiro, H.; Kawaguchi, A.; Aishima, S. Transferrin receptor 1 overexpression is associated with tumour de-differentiation and acts as a potential prognostic indicator of hepatocellular carcinoma. Histopathology 2019, 75, 63–73. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Cui, C.; Cheng, X.; Yan, L.; Ding, H.; Guan, X.; Zhang, W.; Tian, X.; Hao, C. Downregulation of TfR1 promotes progression of colorectal cancer via the JAK/STAT pathway. Cancer Manag. Res. 2019, 11, 6323–6341. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Hwang, S.; Seong, R.H. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem. Biophys. Res. Commun. 2016, 471, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Mitra, S.; Wu, G.; Berka, V.; Song, J.; Yu, Y.; Poget, S.; Wang, D.N.; Tsai, A.L.; Zhou, M. Six-Transmembrane Epithelial Antigen of Prostate 1 (STEAP1) has a single b heme and is capable of reducing metal ion complexes and oxygen. Biochemistry 2016, 55, 6673–6684. [Google Scholar] [CrossRef] [PubMed]
- Whiteland, H.; Spencer-Harty, S.; Morgan, C.; Kynaston, H.; Thomas, D.H.; Bose, P.; Fenn, N.; Lewis, P.; Jenkins, S.; Doak, S.H. A role for STEAP2 in prostate cancer progression. Clin. Exp. Metastasis 2014, 31, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Xu, R.; Wang, S.; Yang, N.; Ni, S.; Zhang, Q.; Xu, Y.; Zhang, X.; Zhang, C.; Wei, Y.; et al. Six-Transmembrane Epithelial Antigen of Prostate 3 Predicts Poor Prognosis and Promotes Glioblastoma Growth and Invasion. Neoplasia 2018, 20, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Burnell, S.E.A.; Spencer-Harty, S.; Howarth, S.; Bodger, O.; Kynaston, H.; Morgan, C.; Doak, S.H. STEAP2 knockdown reduces the invasive potential of prostate cancer cells. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Hann, H.W.; Ye, Z.; Yin, C.; Wang, Y.; Fang, W.; Wan, S.; Wang, C.; Tao, K. Ferritin level prospectively predicts hepatocarcinogenesis in patients with chronic hepatitis B virus infection. Oncol. Lett. 2018, 16, 3499–3508. [Google Scholar] [CrossRef]
- Wang, X.; An, P.; Zeng, J.; Liu, X.; Wang, B.; Fang, X.; Wang, F.; Ren, G.; Min, J. Serum ferritin in combination with prostate-specific antigen improves predictive accuracy for prostate cancer. Oncotarget 2017, 8, 17862–17872. [Google Scholar] [CrossRef]
- Ji, M.; Li, X.D.; Shi, H.B.; Ning, Z.H.; Zhao, W.Q.; Wang, Q.; Zhu, L.N.; Liu, Y.; Wu, C.P. Clinical significance of serum ferritin in elderly patients with primary lung carcinoma. Tumor Biol. 2014, 35, 10195–10199. [Google Scholar] [CrossRef]
- Wang, S.L.; Cao, S.; Wu, R.; Chi, F.; Tang, M.Y.; Jin, X.Y.; Chen, X.D. Serum ferritin predicted prognosis in patients with locally advanced pancreatic cancer. Future Oncol. 2015, 11, 2905–2910. [Google Scholar] [CrossRef]
- Wang, W.; Knovich, M.A.; Coffman, L.G.; Torti, F.M.; Torti, S.V. Serum Ferritin: Past, Present and Future. Biochim. Biophys. Acta 2010, 1800, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.N.; Su, D.; Wang, L.; Yu, F.L. Roles of the hepcidin-ferroportin axis and iron in cancer. Eur. J. Cancer Prev. 2014, 23, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Wei, Z.; Shaikh, Z.A. Suppression of ferroportin expression by cadmium stimulates proliferation, EMT, and migration in triple-negative breast cancer cells. Toxicol. Appl. Pharmacol. 2018, 356, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zhou, C.X.; Shi, Y.B.; Lu, H.; He, X.Z. Decreased expression of ferroportin in prostate cancer. Oncol. Lett. 2015, 10, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Vela, D.; Vela-Gaxha, Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp. Mol. Med. 2018, 50, e436. [Google Scholar] [CrossRef]
- Brookes, M.J.; Hughes, S.; Turner, F.E.; Reynolds, G.; Sharma, N.; Ismail, T.; Berx, G.; McKie, A.T.; Hotchin, N.; Anderson, G.J.; et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006, 55, 1449–1460. [Google Scholar] [CrossRef]
- Josson, S.; Nomura, T.; Lin, J.T.; Huang, W.C.; Wu, D.; Zhau, H.E.; Zayzafoon, M.; Weizmann, M.N.; Gururajan, M.; Chung, L.W. β2-microglobulin induces epithelial to mesenchymal transition and confers cancer lethality and bone metastasis in human cancer cells. Cancer Res. 2011, 71, 2600–2610. [Google Scholar] [CrossRef]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Basso, G.; Marchesi, F.; Doni, A.; Marra, G.; Roncalli, M.; Mantovani, A.; et al. Presence of Twist1-positive neoplastic cells in the stroma of chromosome-unstable colorectal tumors. Gastroenterology 2013, 145, 647–657. [Google Scholar] [CrossRef]
- Troadec, M.B.; Loréal, O.; Brissot, P. The interaction of iron and the genome: For better and for worse. Mutat. Res. 2017, 774, 25–32. [Google Scholar] [CrossRef]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R., Jr.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates HFE-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Alexander, J.; Kowdley, K.V. HFE-associated hereditary hemochromatosis. Genet. Med. 2009, 11, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Valenti, L.; Fracanzani, A.L. Hemochromatosis gene (HFE) mutations and cancer risk: Expanding the clinical manifestations of hereditary iron overload. Hepatology 2010, 51, 1119–1121. [Google Scholar] [CrossRef]
- Cauza, E.; Peck-Radosavljevic, M.; Ulrich-Pur, H.; Datz, C.; Gschwantler, M.; Schöniger-Hekele, M.; Wrba, F.; Hackl, F.; Polli, C.; Gangl, A.; et al. Mutations of the HFE gene in patients with hepatocellular carcinoma. Am. J. Gastroenterol. 2003, 98, 442–447. [Google Scholar] [CrossRef]
- Zhang, M.; Xiong, H.; Fang, L.; Lu, W.; Wu, X.; Wang, Y.Q.; Cai, Z.M.; Wu, S. Meta-Analysis of the Association between H63D and C282Y Polymorphisms in HFE and Cancer Risk. Asian Pac. J Cancer Prev. 2015, 16, 4633–4639. [Google Scholar] [CrossRef]
- Osborne, N.J.; Gurrin, L.C.; Allen, K.J.; Constantine, C.C.; Delatycki, M.B.; McLaren, C.E.; Gertig, D.M.; Anderson, G.J.; Southey, M.C.; Olynyk, J.K.; et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology 2010, 51, 1311–1318. [Google Scholar] [CrossRef]
- Robinson, J.P.; Johnson, V.L.; Rogers, P.A.; Houlston, R.S.; Maher, E.R.; Bishop, D.T.; Evans, D.G.; Thomas, H.J.; Tomlinson, I.P.; Silver, A.R. Evidence for an association between compound heterozygosity for germ line mutations in the hemochromatosis (HFE) gene and increased risk of colorectal cancer. Cancer Epidemiol Biomark. Prev. 2005, 14, 1460–1463. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.F.; Chang, X.; Hua, R.X.; Yan, G.N.; Meng, G.; Liao, X.Y.; Zhang, X.; Guo, Q.N. The risk of new-onset cancer associated with HFE C282Y and H63D mutations: Evidence from 87,028 participants. J. Cell. Mol. Med. 2016, 20, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, H.; Li, T.; Yao, H. HFE gene C282Y variant is associated with colorectal cancer in Caucasians: A meta-analysis. Tumor Biol. 2013, 34, 2255–2259. [Google Scholar] [CrossRef] [PubMed]
- Agudo, A.; Bonet, C.; Sala, N.; Muñoz, X.; Aranda, N.; Fonseca-Nunes, A.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; Vineis, P.; Panico, S.; et al. Hemochromatosis (HFE) gene mutations and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Carcinogenesis 2013, 34, 1244–1250. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, C.; Hu, M.; Li, J.; Liu, R. Plasma ferritin levels, HFE polymorphisms, and risk of pancreatic cancer among Chinese Han population. Tumor Biol. 2014, 35, 7629–7633. [Google Scholar] [CrossRef]
- Hucl, T.; Kylanpää-Bäck, M.L.; Witt, H.; Künzli, B.; Lempinen, M.; Schneider, A.; Kemppainen, E.; Löhr, M.; Friess, H.; Ockenga, J.; et al. HFE genotypes in patients with chronic pancreatitis and pancreatic adenocarcinoma. Genet. Med. 2007, 9, 479–483. [Google Scholar] [CrossRef]
- Shen, L.L.; Gu, D.Y.; Zhao, T.T.; Tang, C.J.; Xu, Y.; Chen, J.F. Implicating the H63D polymorphism in the HFE gene in increased incidence of solid cancers: A meta-analysis. Genet. Mol. Res. 2015, 14, 13735–13745. [Google Scholar] [CrossRef]
- Lee, S.Y.; Zhu, J.; Salzberg, A.C.; Zhang, B.; Liu, D.J.; Muscat, J.E.; Langan, S.T.; Connor, J.R. Analysis of single nucleotide variants of HFE gene and association to survival in The Cancer Genome Atlas GBM data. PLoS ONE 2017, 12, e0174778. [Google Scholar] [CrossRef]
- Laghi, L.; Malesci, A. Microsatellite instability and therapeutic consequences in colorectal cancer. Dig. Dis. 2012, 30, 304–309. [Google Scholar] [CrossRef]
- Shi, Z.; Johnstone, D.; Talseth-Palmer, B.A.; Evans, T.J.; Spigelman, A.D.; Groombridge, C.; Milward, E.A.; Olynyk, J.K.; Suchy, J.; Kurzawski, G.; et al. Haemochromatosis HFE gene polymorphisms as potential modifiers of hereditary nonpolyposis colorectal cancer risk and onset age. Int. J. Cancer 2009, 125, 78–83. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181, 219–222. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of cytochrome P450S in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef]
- Rhee, S.G.; Yang, K.S.; Kang, S.W.; Woo, H.A.; Chang, T.S. Controlled elimination of intracellular H(2)O(2): Regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid. Redox Signal. 2005, 7, 619–626. [Google Scholar] [CrossRef]
- Florean, C.; Song, S.; Dicato, M.; Diederich, M. Redox biology of regulated cell death in cancer: A focus on necroptosis and ferroptosis. Free Radic. Biol. Med. 2019, 134, 177–189. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Li Volti, G.; Tibullo, D.; Vanella, L.; Giallongo, C.; Di Raimondo, F.; Forte, S.; Di Rosa, M.; Signorelli, S.S.; Barbagallo, I. The Heme Oxygenase System in Hematological Malignancies. Antioxid. Redox Signal. 2017, 27, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.F.; Yeh, C.T.; Sun, Y.J.; Chiang, M.T.; Lan, W.M.; Li, F.A.; Chau, L.Y.; Lee, W.H. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 2014. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, S.; Kim, E.J.; Park, J.H.; Yang, S.H.; Jeong, E.T.; Park, C.; Youn, M.J.; So, H.S.; Park, R. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer 2008, 60, 47–56. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177. [Google Scholar]
- Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1(HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef]
- Miyake, M.; Fujimoto, K.; Anai, S.; Ohnishi, S.; Nakai, Y.; Inoue, T.; Matsumura, Y.; Tomioka, A.; Ikeda, T.; Okajima, E. Inhibition of heme oxygenase-1 enhances the cytotoxic effect of gemcitabine in urothelial cancer cells. Anticancer Res. 2010, 30, 2145–2152. [Google Scholar]
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Künzli, B.; Autschbach, F.; Meuer, S.; Büchler, M.W.; Friess, H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005, 11, 3790–3798. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Ahmad, I.M.; Rachagani, S.; Banerjee, K.; Thompson, C.M.; Maurer, H.C.; Olive, K.P.; Bailey, K.L.; Britigan, B.E.; Kumar, S. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl. Res. 2019, 207, 56–69. [Google Scholar] [CrossRef]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Filardi, E.; Vega, M.A.; Sánchez-Mateos, P.; Corbí, A.L.; Puig-Kröger, A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Iriki, T.; Tsuboki, J.; Cheng, P.; Nakagata, N.; Mizuta, H.; Bekki, H.; et al. CD163 Is Required for Protumoral Activation of Macrophages in Human and Murine Sarcoma. Cancer Res. 2018, 78, 3255–3266. [Google Scholar] [CrossRef]
- Guadagno, E.; Presta, I.; Maisano, D.; Donato, A.; Pirrone, C.K.; Cardillo, G.; Corrado, S.D.; Mignogna, C.; Mancuso, T.; Donato, G.; et al. Role of Macrophages in Brain Tumor Growth and Progression. Int. J. Mol. Sci. 2018, 19, 1005. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 2017, 6, e1342918. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Kukulj, S.; Jaganjac, M.; Boranic, M.; Krizanac, S.; Santic, Z.; Poljak-Blazi, M. Altered iron metabolism, inflammation, transferrin receptors, and ferritin expression in non-small-cell lung cancer. Med. Oncol. 2010, 27, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Chung, J.W.; Lapointe, R.; Santos, M.M. The hemochromatosis protein HFE 20 years later: An emerging role in antigen presentation and in the immune system. Immun. Inflamm. Dis. 2017, 5, 218–232. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems bunderstanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Salter-Cid, L.; Peterson, P.A.; Yang, Y. The major histocompatibility complex-encoded HFE in iron homeostasis and immune function. Immunol. Res. 2000, 22, 43–59. [Google Scholar] [CrossRef]
- Pascolo, S.; Ginhoux, F.; Laham, N.; Walter, S.; Schoor, O.; Probst, J.; Rohrlich, P.; Obermayr, F.; Fisch, P.; Danos, O.; et al. The non-classical HLA class I molecule HFE does not influence the NK-like activity contained in fresh human PBMCs and does not interact with NK cells. Int. Immunol. 2005, 17, 117–122. [Google Scholar] [CrossRef]
- Porto, G.; Cruz, E.; Teles, M.J.; De Sousa, M. HFE Related Hemochromatosis: Uncovering the inextricable link between iron homeostasis and the immunological system. Pharmaceuticals 2019, 12, 122. [Google Scholar] [CrossRef]
- Costa, M.; Cruz, E.; Oliveira, S.; Benes, V.; Ivacevic, T.; Silva, M.J.; Vieira, I.; Dias, F.; Fonseca, S.; Gonçalves, M.; et al. Lymphocyte gene expression signatures from patients and mouse models of hereditary hemochromatosis reveal a function of HFE as a negative regulator of CD8+ T-lymphocyte activation and differentiation in vivo. PLoS ONE 2015, 10, e0124246. [Google Scholar] [CrossRef]
- Reuben, A.; Phénix, M.; Santos, M.M.; Lapointe, R. The WT hemochromatosis protein HFE inhibits CD8⁺ T-lymphocyte activation. Eur. J. Immunol. 2014, 44, 1604–1614. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron Metabolism in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2257. https://doi.org/10.3390/ijms21062257
Forciniti S, Greco L, Grizzi F, Malesci A, Laghi L. Iron Metabolism in Cancer Progression. International Journal of Molecular Sciences. 2020; 21(6):2257. https://doi.org/10.3390/ijms21062257
Chicago/Turabian StyleForciniti, Stefania, Luana Greco, Fabio Grizzi, Alberto Malesci, and Luigi Laghi. 2020. "Iron Metabolism in Cancer Progression" International Journal of Molecular Sciences 21, no. 6: 2257. https://doi.org/10.3390/ijms21062257
APA StyleForciniti, S., Greco, L., Grizzi, F., Malesci, A., & Laghi, L. (2020). Iron Metabolism in Cancer Progression. International Journal of Molecular Sciences, 21(6), 2257. https://doi.org/10.3390/ijms21062257