Effects of High-Dose Ionizing Radiation in Human Gene Expression: A Meta-Analysis

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
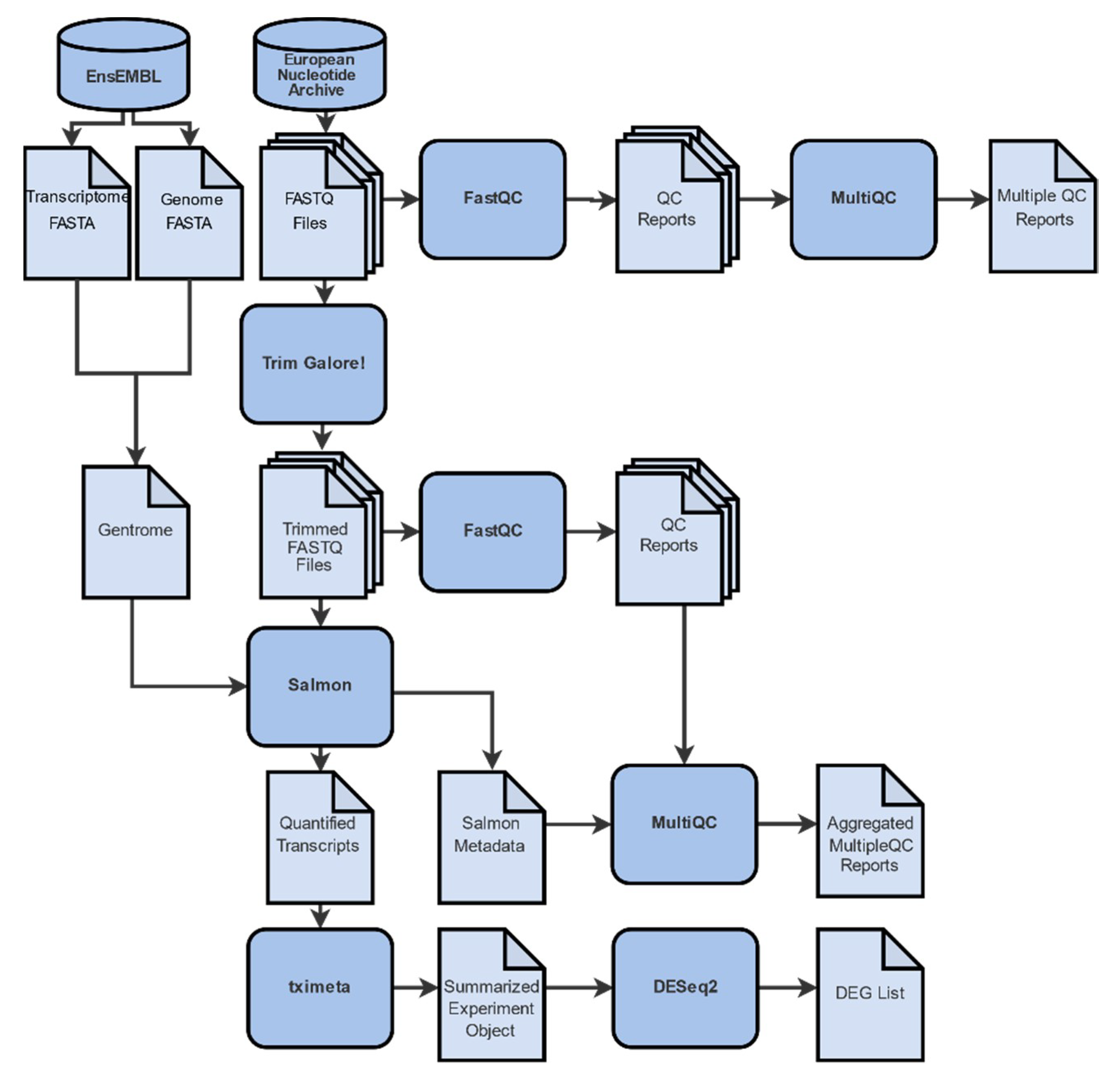
2.1. Data Collection, Filtering, Pre-Processing and Mapping
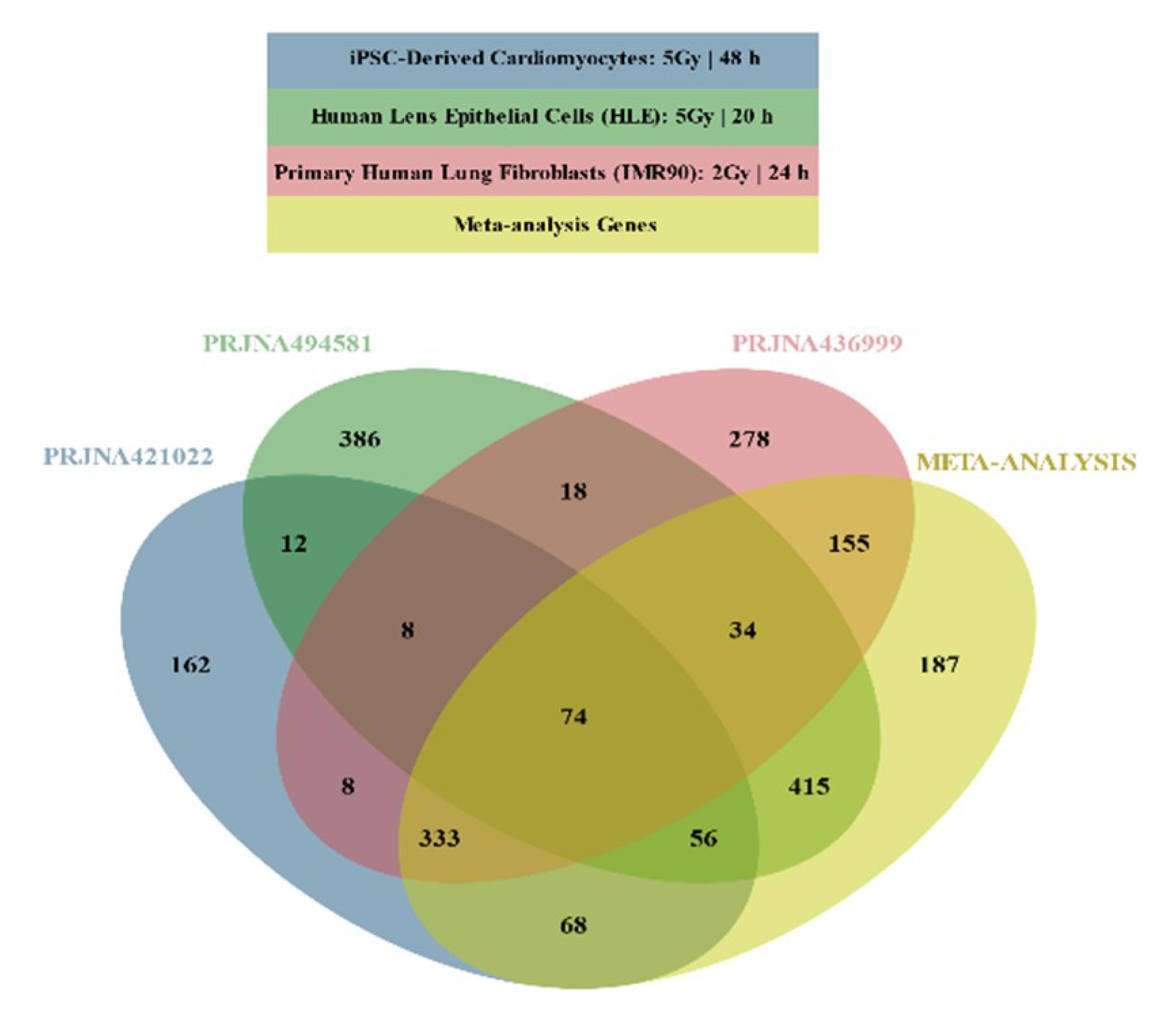
2.2. Differential Gene Expression
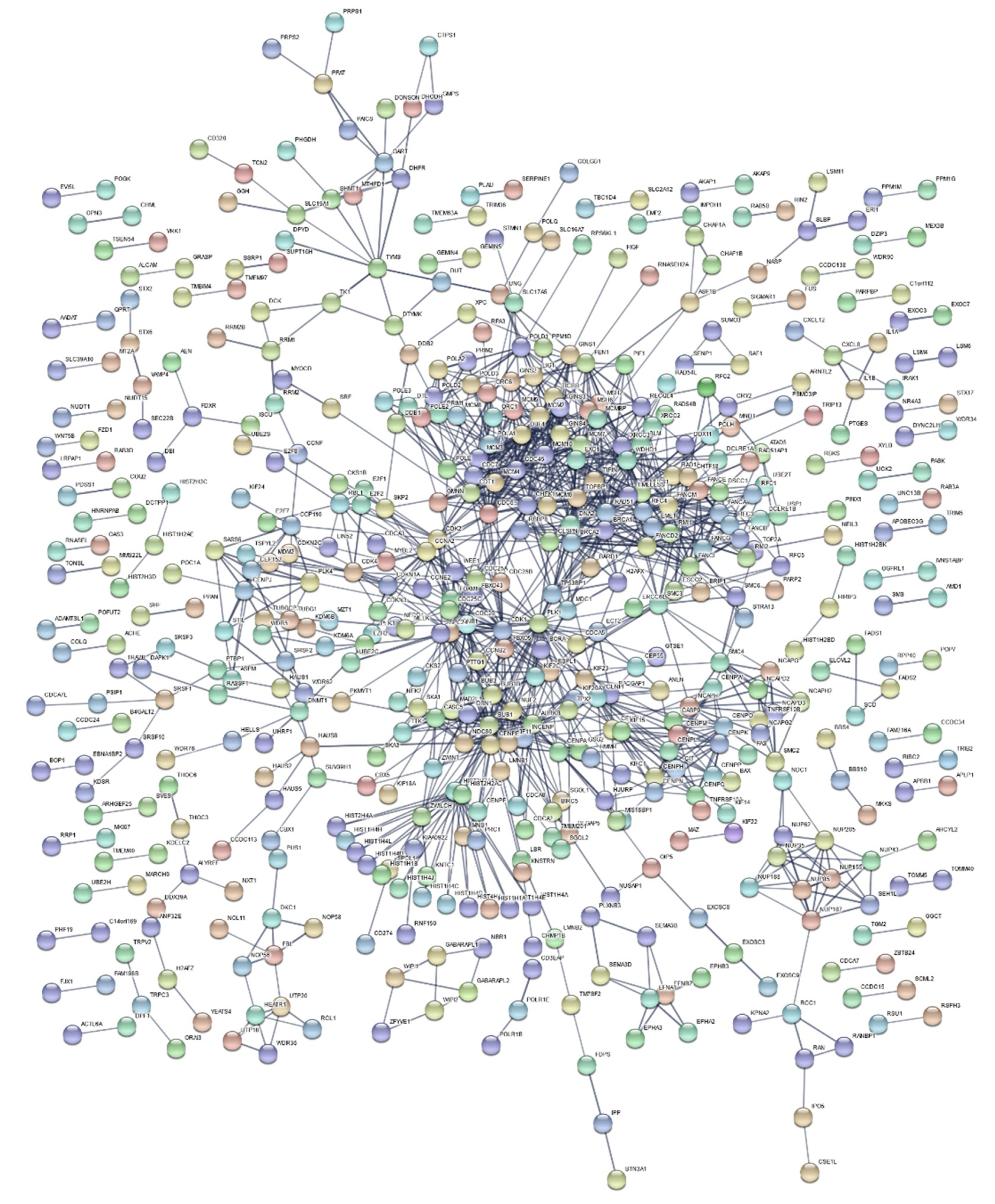
2.3. Functional Enrichment Results
3. Discussion
4. Materials and Methods
4.1. Datasets
4.2. Raw Read Evaluation
4.3. Sequence Alignment
4.4. Transcript Quantification
4.5. Differential Gene Expression Analysis
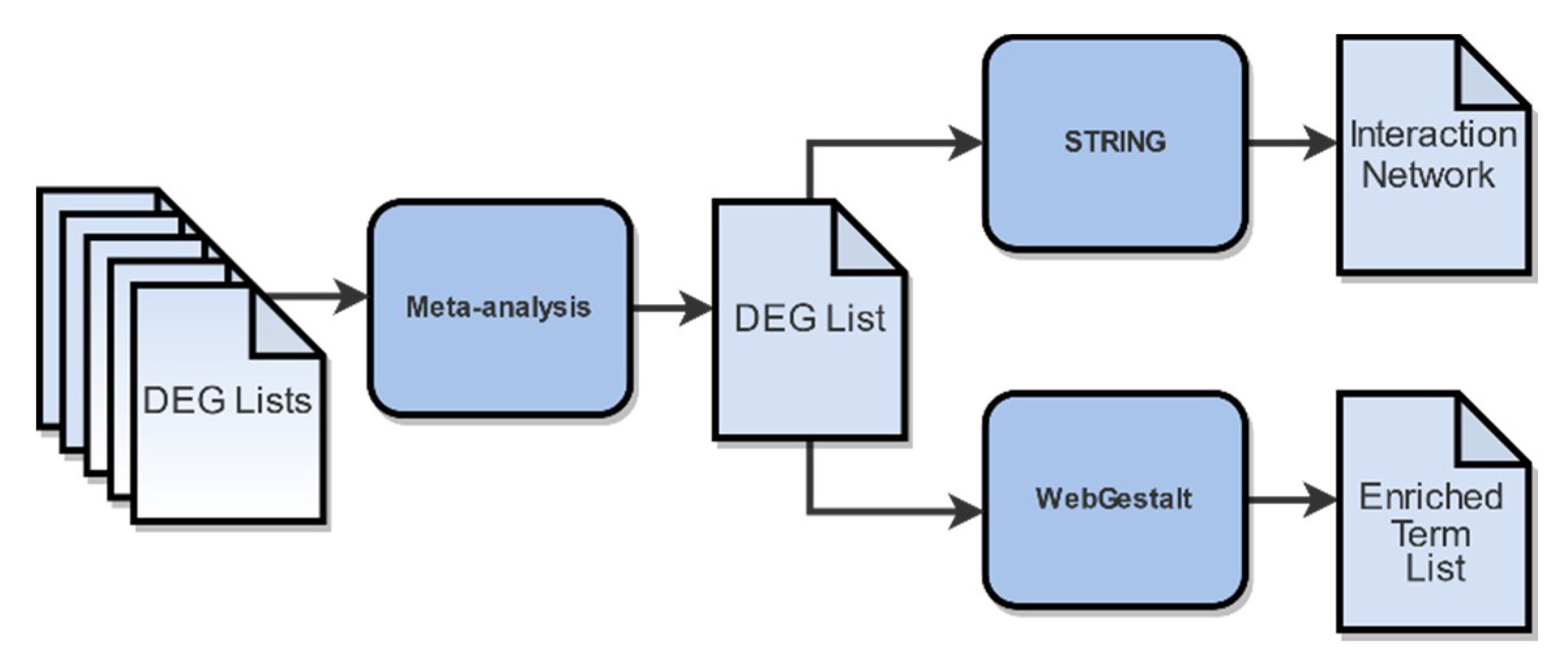
4.6. Meta-Analysis
4.7. Functional Enrichment Analysis and Gene Network Construction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BER | Base Excision Repair |
| DDR | DNA Damage Response |
| DEGs | Differentially Expressed Genes |
| DGE | Differential Gene Expression |
| DGEA | Differential Gene Expression Analysis |
| DSBs | Double Strand Breaks |
| ENA | European Nucleotide Archive |
| FDR | False Discovery Rate |
| GSEA | Gene Set Enrichment Analysis |
| GO | Gene Ontology |
| HR | Homology-dependent Recombination |
| IR | Ionizing Radiation |
| LET | Linear Energy Transfer |
| Log2FC | Log2 Fold Change |
| MMR | Mismatch Repair |
| NER | Nucleotide Excision Repair |
| NGS | Next-Generation Sequencing |
| NHEJ | Non-Homologous End Joining |
| ORA | Over-Representation Analysis |
| PPI | Protein to Protein Interaction |
| Q | PHRED Score |
| RBE | Relative Biological Effectiveness |
| RNA-Seq | RNA-Sequencing |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| RT | Radiation Therapy |
| SSBs | Single-Strand Breaks |
| WebGestalt | WEB-based Gene Set Analysis Toolkit |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trim Galore! | trim_galore --cores 4 --illumina -q 20 --phred33 –paired --fastqc <fastq files> |
| Salmon | salmon quant -i salmon_index --libType A -1 <forward_1.fq.gz> -2 <reverse_1.fq.gz> --gcBias --validateMappings -o <transcripts_directory> |
| WebGestalt | Basic Parameters: |
| Organism of Interest: Homo sapiens Method of Interest: ORA Functional Database: geneontology + (Biological Process: no redundant), pathway + (KEGG), Network + (Transcription Factor target) Gene List: Select Gene ID Type: EnsEMBL Gene IDReference Gene List: Upload: Mappings per study: EnsEMBL Gene ID | |
| Advanced Parameters | |
| minimum number of genes for category: 2 Multiple Test Adjustment: Benjamini-Hochberg Significance level: FDR (0.05) Number of categories visualized in the report: 100 | |
| STRING | Basic Settings: |
| meaning of network edges: confidence active interaction sources: textmining minimum required interaction score: high confidence (0.7) | |
| Advanced Settings: | |
| hide disconnected nodes in the network disable structure previous inside network bubbles |
References
- UNSCEAR (United Nations Scientific Committee on the Effects of Atomic Radiation). Biological Mechanisms of Radiation Actions at Low Doses; United Nations: New York, NY, USA, 2012. [Google Scholar]
- Leuraud, K.; Richardson, D.B.; Cardis, E.; Daniels, R.D.; Gillies, M.; O’Hagan, J.A.; Hamra, G.B.; Haylock, R.; Laurier, D.; Moissonnier, M.; et al. Ionising radiation and risk of death from leukaemia and lymphoma in radiation-monitored workers (INWORKS): An international cohort study. Lancet. Haematol. 2015, 2, e276–e281. [Google Scholar] [CrossRef]
- Ray, M.; Yunis, R.; Chen, X.; Rocke, D.M. Comparison of low and high dose ionising radiation using topological analysis of gene coexpression networks. Bmc Genom. 2012, 13, 190. [Google Scholar] [CrossRef]
- Bragg, W.H.; Kleeman, R. LXXIV. On the ionization curves of radium. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1904, 8, 726–738. [Google Scholar] [CrossRef]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef]
- Mehta, S.R.; Suhag, V.; Semwal, M.; Sharma, N. Radiotherapy: Basic Concepts and Recent Advances. Med. J. Armed. Forces India 2010, 66, 158–162. [Google Scholar] [CrossRef]
- Kjellberg, R.N.; Hanamura, T.; Davis, K.R.; Lyons, S.L.; Adams, R.D. Bragg-Peak Proton-Beam Therapy for Arteriovenous Malformations of the Brain. N. Engl. J. Med. 1983, 309, 269–274. [Google Scholar] [CrossRef]
- Bernier, J.; Hall, E.J.; Giaccia, A. Radiation oncology: A century of achievements. Nat. Rev. Cancer 2004, 4, 737–747. [Google Scholar] [CrossRef]
- Georgakilas, A.G. Bystander and non-targeted effects: A unifying model from ionizing radiation to cancer. Cancer Lett. 2015, 356, 3–4. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Wallace, S.S. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat. Res. 1998, 150, S60–S79. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef]
- Georgakilas, A.G. Processing of DNA damage clusters in human cells: Current status of knowledge. Mol. Biosyst. 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef]
- Saini, D.; Shelke, S.; Mani Vannan, A.; Toprani, S.; Jain, V.; Das, B.; Seshadri, M. Transcription profile of DNA damage response genes at G(0) lymphocytes exposed to gamma radiation. Mol. Cell. Biochem. 2012, 364, 271–281. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Pavlopoulou, A.; Hola, M.; Dona, M.; Michalopoulos, I.; Balestrazzi, A.; Angelis, K.J.; Georgakilas, A.G. Bridging Plant and Human Radiation Response and DNA Repair through an In Silico Approach. Cancers 2017, 9, 65. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254 e236. [Google Scholar] [CrossRef]
- Friedberg, E.C. A history of the DNA repair and mutagenesis field: The discovery of base excision repair. Dna Repair 2016, 37, A35–A39. [Google Scholar] [CrossRef]
- Pan, M.R.; Li, K.; Lin, S.Y.; Hung, W.C. Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci. 2016, 17, 685. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Pavlopoulou, A.; Louka, M.; Nikitaki, Z.; Vorgias, C.E.; Bagos, P.G.; Michalopoulos, I. Emerging molecular networks common in ionizing radiation, immune and inflammatory responses by employing bioinformatics approaches. Cancer Lett. 2015. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef]
- Hatzi, V.I.; Laskaratou, D.A.; Mavragani, I.V.; Nikitaki, Z.; Mangelis, A.; Panayiotidis, M.I.; Pantelias, G.E.; Terzoudi, G.I.; Georgakilas, A.G. Non-targeted radiation effects in vivo: A critical glance of the future in radiobiology. Cancer Lett. 2015, 356, 34–42. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. Plos ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip Rev. Rna. 2017, 8. [Google Scholar] [CrossRef]
- Romero, J.P.; Ortiz-Estevez, M.; Muniategui, A.; Carrancio, S.; de Miguel, F.J.; Carazo, F.; Montuenga, L.M.; Loos, R.; Pio, R.; Trotter, M.W.B.; et al. Comparison of RNA-seq and microarray platforms for splice event detection using a cross-platform algorithm. Bmc Genom. 2018, 19, 703. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Chauhan, V.; Rowan-Carroll, A.; Gagne, R.; Kuo, B.; Williams, A.; Yauk, C.L. The use of in vitro transcriptional data to identify thresholds of effects in a human lens epithelial cell-line exposed to ionizing radiation. Int. J. Radiat. Biol. 2019, 95, 156–169. [Google Scholar] [CrossRef]
- Shimada, M.; Tsukada, K.; Kagawa, N.; Matsumoto, Y. Reprogramming and differentiation-dependent transcriptional alteration of DNA damage response and apoptosis genes in human induced pluripotent stem cells. J. Radiat. Res. 2019, 60, 719–728. [Google Scholar] [CrossRef]
- Becker, B.V.; Majewski, M.; Abend, M.; Palnek, A.; Nestler, K.; Port, M.; Ullmann, R. Gene expression changes in human iPSC-derived cardiomyocytes after X-ray irradiation. Int. J. Radiat. Biol. 2018, 94, 1095–1103. [Google Scholar] [CrossRef]
- Becker, B.V.; Kaatsch, L.; Obermair, R.; Schrock, G.; Port, M.; Ullmann, R. X-ray irradiation induces subtle changes in the genome-wide distribution of DNA hydroxymethylation with opposing trends in genic and intergenic regions. Epigenetics 2019, 14, 81–93. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Schurch, N.J.; Schofield, P.; Gierlinski, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef]
- Liao, Y.; Shi, W. Read trimming is not required for mapping and quantification of RNA-seq reads. bioRxiv 2019. [Google Scholar] [CrossRef]
- Williams, C.R.; Baccarella, A.; Parrish, J.Z.; Kim, C.C. Trimming of sequence reads alters RNA-Seq gene expression estimates. Bmc Bioinform. 2016, 17, 103. [Google Scholar] [CrossRef]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb Protoc 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Krizanovic, K.; Echchiki, A.; Roux, J.; Sikic, M. Evaluation of tools for long read RNA-seq splice-aware alignment. Bioinformatics 2018, 34, 748–754. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R.; International Nucleotide Sequence Database, C. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Belinky, F.; Nativ, N.; Stelzer, G.; Zimmerman, S.; Iny Stein, T.; Safran, M.; Lancet, D. PathCards: Multi-source consolidation of human biological pathways. J. Biol. Databases Curation 2015, 2015. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Shu, K.X.; Li, B.; Wu, L.X. The p53 network: p53 and its downstream genes. Colloids Surf. Biointerfaces 2007, 55, 10–18. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.; Huna, A.; Warnier, M.; Flaman, J.M.; Bernard, D. Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell Death Dis. 2018, 9, 259. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Greene, M.W. Circadian rhythms and tumor growth. Cancer Lett. 2012, 318, 115–123. [Google Scholar] [CrossRef]
- Forssell-Aronsson, E.; Quinlan, R.A. The Impact of Circadian Rhythms on Medical Imaging and Radiotherapy Regimes for the Paediatric Patient. Radiat. Prot. Dosim. 2017, 173, 16–20. [Google Scholar] [CrossRef]
- Jim, H.S.; Lin, H.Y.; Tyrer, J.P.; Lawrenson, K.; Dennis, J.; Chornokur, G.; Chen, Z.; Chen, A.Y.; Permuth-Wey, J.; Aben, K.K.; et al. Common Genetic Variation in Circadian Rhythm Genes and Risk of Epithelial Ovarian Cancer (EOC). J. Genet. Genome Res. 2015, 2. [Google Scholar] [CrossRef]
- Kizaki, T.; Sato, S.; Shirato, K.; Sakurai, T.; Ogasawara, J.; Izawa, T.; Ohira, Y.; Suzuki, K.; Ohno, H. Effect of Circadian Rhythm on Clinical and Pathophysiological Conditions and Inflammation. Crit. Rev. Immunol. 2015, 35, 261–275. [Google Scholar] [CrossRef]
- Silvester, N.; Alako, B.; Amid, C.; Cerdeno-Tarraga, A.; Clarke, L.; Cleland, I.; Harrison, P.W.; Jayathilaka, S.; Kay, S.; Keane, T.; et al. The European Nucleotide Archive in 2017. Nucleic Acids Res. 2018, 46, D36–D40. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 March 2020).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Krueger, F. Trim Galore!: A Wrapper Tool around Cutadapt and Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files, with Some Extra Functionality for MspI-Digested RRBS-Type (Reduced Representation Bisufite-Seq) Libraries. 2015. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 10 March 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Srivastava, A.; Malik, L.; Sarkar, H.; Zakeri, M.; Almodaresi, F.; Soneson, C.; Love, M.I.; Kingsford, C.; Patro, R. Alignment and mapping methodology influence transcript abundance estimation. bioRxiv 2019. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2019. Available online: https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing (accessed on 10 March 2020).
- RStudio Team. RStudio: Integrated Development for R. RstudioInc. 2019. Available online: https://rstudio.com/ (accessed on 10 March 2020).
- Love, M.I.; Soneson, C.; Charlotte, H.; Johnson, L.; Pierce, N.; Shepherd, L.; Morgan, M.; Patro, R. Tximeta: Reference sequence checksums for provenance identification in RNA-seq. PLOS Comput. Biol. 2019. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human. 2019. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 10 March 2020).
- Yates, B.; Braschi, B.; Gray, K.A.; Seal, R.L.; Tweedie, S.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2017. Nucleic Acids Res. 2017, 45, D619–D625. [Google Scholar] [CrossRef]
- Stouffer, S.A.; Suchman, E.A.; DeVinney, L.C.; Star, S.A.; Williams, R.M. The American Soldier: Adjustment During Army Life; Princeton University Press: Princeton, NJ, USA, 1949; p. 1. [Google Scholar]
- Mosteller, F.; Bush, R.R. Selected quantitative techniques. In Handbook of social psychology: Vol. 1. Theory and Method; Lindzey, G., Ed.; Addison-Wesley: Cambridge, UK, 1954; pp. 289–334. [Google Scholar]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. Plos Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]




| Bioproject | Geo | IR Type | Tissue | Condition | Sample Count |
|---|---|---|---|---|---|
| PRJNA494581 | GSE120805 | X-rays | Human Lens Epithelial Cells | Control | 5 |
| 2 Gy | 20 h | 5 | ||||
| 5 Gy | 20 h | 5 | ||||
| PRJNA421022 | GSE107685 | X-rays | iPSC-Derived Cardiomyocytes | Control | 3 |
| 5 Gy| 48 h | 3 | ||||
| PRJNA436999 | GSE111437 | X-rays | Primary Human Lung Fibroblasts (IMR90) | Control | 6 h | 3 |
| Control | 24 h | 3 | ||||
| 2 Gy | 6 h | 3 | ||||
| 2 Gy | 24 h | 3 | ||||
| PRJNA396832 | GSE102145 | X-rays | Skin Fibroblasts (WS1) | Control | 1 |
| 5 Gy | 24 h | 1 | ||||
| PRJNA450083 | GSE113125 | Gamma-rays | Skin Fibroblasts | Control | 1 |
| 5 Gy | 1 h | 1 | ||||
| iPSC-Fibroblasts | Control | 1 | |||
| 5 Gy | 1 h | 1 | ||||
| iPSC-Neural Progenitor Cells | Control | 1 | |||
| 5 Gy | 1 h | 1 |
| Bioproject Accession | PRJNA421022 | PRJNA436999 | PRJNA494581 | ||
|---|---|---|---|---|---|
| GEO Accession | GSE107685 | GSE111437 | GSE120805 | ||
| IR Type | X-rays | X-rays | X-rays | ||
| Cell Type | iPSC-Derived Cardiomyocytes | Primary Human Lung Fibroblasts (IMR90) | Human Lens Epithelial Cells (HLE) | ||
| Dose | 5 Gy | 2 Gy | 2 Gy | 2 Gy | 5 Gy |
| Time Point | 48 h | 6 h | 24 h | 20 h | 20 h |
| DEG Counts | 721 | 353 | 908 | 59 | 1003 |
| Gene Set | Description | Source | FDR |
|---|---|---|---|
Up-Regulated Genes | |||
| GO:0072331 | Signal transduction by p53 class mediator | GO | 0.0022959 |
| hsa04115 | p53 signaling pathway | KEGG | 2.24 × 10−9 |
| GO:0042770 | Signal transduction in response to DNA damage | GO | 0.009620632 |
| GO:0097193 | Intrinsic apoptotic signaling pathway | GO | 0.010992521 |
| GO:0071496 | Cellular response to external stimulus | GO | 0.01930771 |
| GO:0104004 | Cellular response to environmental stimulus | GO | 0.032524732 |
| GO:0008643 | Carbohydrate transport | GO | 0.037630683 |
| hsa01524 | Platinum drug resistance | KEGG | 0.032524732 |
Down-Regulated Genes | |||
| GO:0000075 | Cell cycle checkpoint | GO | 0 |
| hsa04110 | Cell cycle | KEGG | 0 |
| GO:0051321 | Meiotic cell cycle | GO | 0 |
| GO:0044772 | Mitotic cell cycle phase transition | GO | 0 |
| GO:0045930 | Negative regulation of mitotic cell cycle | GO | 0 |
| GO:1902850 | Microtubule cytoskeleton organization involved in mitosis | GO | 0 |
| GO:0044839 | Cell cycle G2/M phase transition | GO | 0 |
| GO:0044843 | Cell cycle G1/S phase transition | GO | 0 |
| GO:1901987 | Regulation of cell cycle phase transition | GO | 0 |
| GO:0010948 | Negative regulation of cell cycle process | GO | 0 |
| GO:0045787 | Positive regulation of cell cycle | GO | 0 |
| GO:0007050 | Cell cycle arrest | GO | 7.77 × 10−4 |
| hsa04115 | p53 signaling pathway | KEGG | 0.002014026 |
| GO:0006260 | DNA replication | GO | 0 |
| hsa03030 | DNA replication | KEGG | 0 |
| hsa00240 | Pyrimidine metabolism | KEGG | 5.67 × 10−10 |
| hsa00230 | Purine metabolism | KEGG | 4.39 × 10−5 |
| GO:0042769 | DNA damage response, detection of DNA damage | GO | 1.88 × 10−6 |
| GO:0006310 | DNA recombination | GO | 0 |
| GO:0006302 | Double-strand break repair | GO | 0 |
| GO:0036297 | Inter-strand cross-link repair | GO | 1.35 × 10−12 |
| hsa03430 | Mismatch repair | KEGG | 3.11 × 10−11 |
| hsa03440 | Homologous recombination | KEGG | 3.11 × 10−11 |
| GO:0006284 | Base excision repair | GO | 6.54 × 10−6 |
| hsa03410 | Base excision repair | KEGG | 1.91 × 10−7 |
| GO:0006298 | Mismatch repair | GO | 4.02 × 10−4 |
| GO:0006289 | Nucleotide excision repair | GO | 5.10 × 10−4 |
| hsa03420 | Nucleotide excision repair | KEGG | 2.91 × 10−6 |
| GO:0006333 | Chromatin assembly or disassembly | GO | 0 |
| GO:0007051 | Spindle organization | GO | 0 |
| GO:0071103 | DNA conformation change | GO | 0 |
| GO:0007059 | Chromosome segregation | GO | 0 |
| GO:0061641 | CENP-A containing chromatin organization | GO | 0 |
| GO:0048285 | Organelle fission | GO | 0 |
| GO:0051052 | Regulation of DNA metabolic process | GO | 0 |
| GO:0071824 | Protein-DNA complex subunit organization | GO | 0 |
| hsa03013 | RNA transport | KEGG | 5.66 × 10−5 |
| hsa03008 | Ribosome biogenesis in eukaryotes | KEGG | 4.13 × 10−5 |
| hsa04114 | Oocyte meiosis | KEGG | 0.001082137 |
| hsa04914 | Progesterone-mediated oocyte maturation | KEGG | 0.008441906 |
| hsa05322 | Systemic lupus erythematosus | KEGG | 0 |
| hsa05203 | Viral carcinogenesis | KEGG | 1.46 × 10−11 |
| hsa05206 | MicroRNAs in cancer | KEGG | 0.008110817 |
| hsa03460 | Fanconi anemia pathway | KEGG | 7.71 × 10−11 |
| hsa05166 | Human T-cell leukemia virus 1 infection | KEGG | 5.47 × 10−4 |
| hsa04217 | Necroptosis | KEGG | 1.19 × 10−4 |
| hsa04218 | Cellular senescence | KEGG | 0.0014615 |
| Gene Set | Gene Symbol | Gene Name | FDR |
|---|---|---|---|
| Down-Regulated Genes | |||
| E2F_Q3_01;E2F_Q4_01; E2F_Q6_01;E2F1_Q4_01 | TFDP1 | Transcription factor Dp-1 | 0 |
| E2F1_Q3;E2F1_Q6; E2F1_Q6_01 | E2F1 | E2F transcription factor 1 | 0 |
| E2F1DP1_01 | E2F1;TFDP1 | E2F transcription factor 1; transcription factor Dp-1 | 0 |
| E2F1DP1RB_01 | E2F1;TFDP1; RB1 | E2F transcription factor 1; transcription factor Dp-1; RB transcriptional corepressor 1 | 0 |
| E2F1DP2_01 | TFDP2 | Transcription factor Dp-2 | 0 |
| E2F4DP1_01 | E2F4;TFDP1 | E2F transcription factor 4; transcription factor Dp-1 | 0 |
| E2F4DP2_01 | E2F4;TFDP2 | E2F transcription factor 4; transcription factor Dp-2 | 0 |
| E2F1_Q4; | E2F1 | E2F transcription factor 1 | 9.80 × 10−12 |
| E2F1_Q3_01 | E2F1 | E2F transcription factor 1 | 4.39 × 10−5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanakoglou, D.S.; Michalettou, T.-D.; Vasileiou, C.; Gioukakis, E.; Maneta, D.; Kyriakidis, K.V.; Georgakilas, A.G.; Michalopoulos, I. Effects of High-Dose Ionizing Radiation in Human Gene Expression: A Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 1938. https://doi.org/10.3390/ijms21061938
Kanakoglou DS, Michalettou T-D, Vasileiou C, Gioukakis E, Maneta D, Kyriakidis KV, Georgakilas AG, Michalopoulos I. Effects of High-Dose Ionizing Radiation in Human Gene Expression: A Meta-Analysis. International Journal of Molecular Sciences. 2020; 21(6):1938. https://doi.org/10.3390/ijms21061938
Chicago/Turabian StyleKanakoglou, Dimitrios S., Theodora-Dafni Michalettou, Christina Vasileiou, Evangelos Gioukakis, Dorothea Maneta, Konstantinos V. Kyriakidis, Alexandros G. Georgakilas, and Ioannis Michalopoulos. 2020. "Effects of High-Dose Ionizing Radiation in Human Gene Expression: A Meta-Analysis" International Journal of Molecular Sciences 21, no. 6: 1938. https://doi.org/10.3390/ijms21061938
APA StyleKanakoglou, D. S., Michalettou, T.-D., Vasileiou, C., Gioukakis, E., Maneta, D., Kyriakidis, K. V., Georgakilas, A. G., & Michalopoulos, I. (2020). Effects of High-Dose Ionizing Radiation in Human Gene Expression: A Meta-Analysis. International Journal of Molecular Sciences, 21(6), 1938. https://doi.org/10.3390/ijms21061938