Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein

 , , , , , ,
, , , , , ,
Abstract
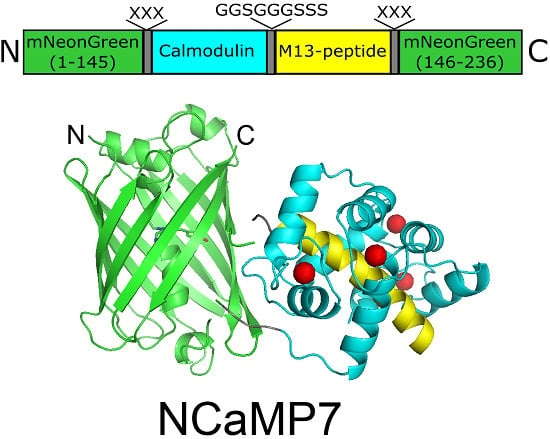
1. Introduction
2. Results and Discussion
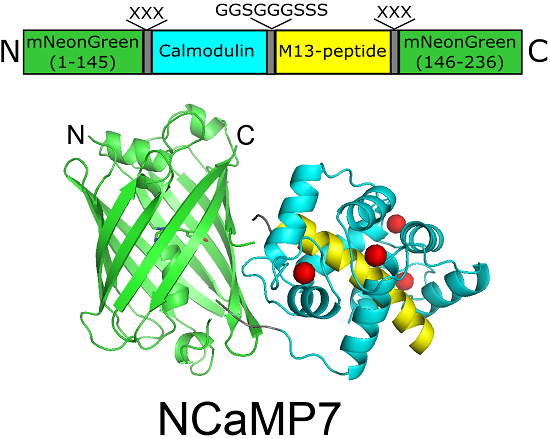
2.1. Development of Bright Green Fluorescent Positive Calcium Indicators Based on the mNeonGreen Protein
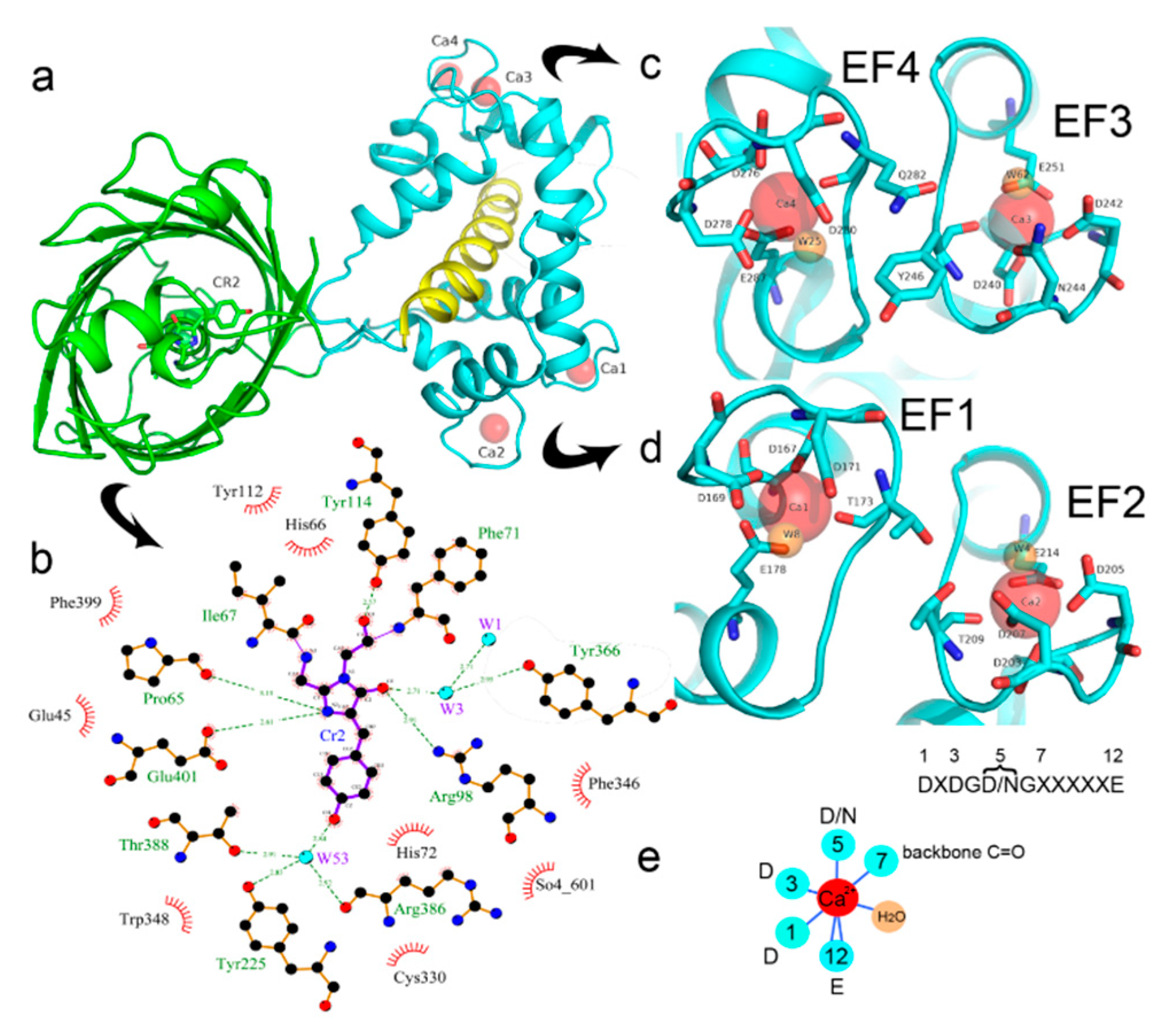
2.2. Crystal Structure of the NCaMP7 Indicator in Its Calcium-Saturated State
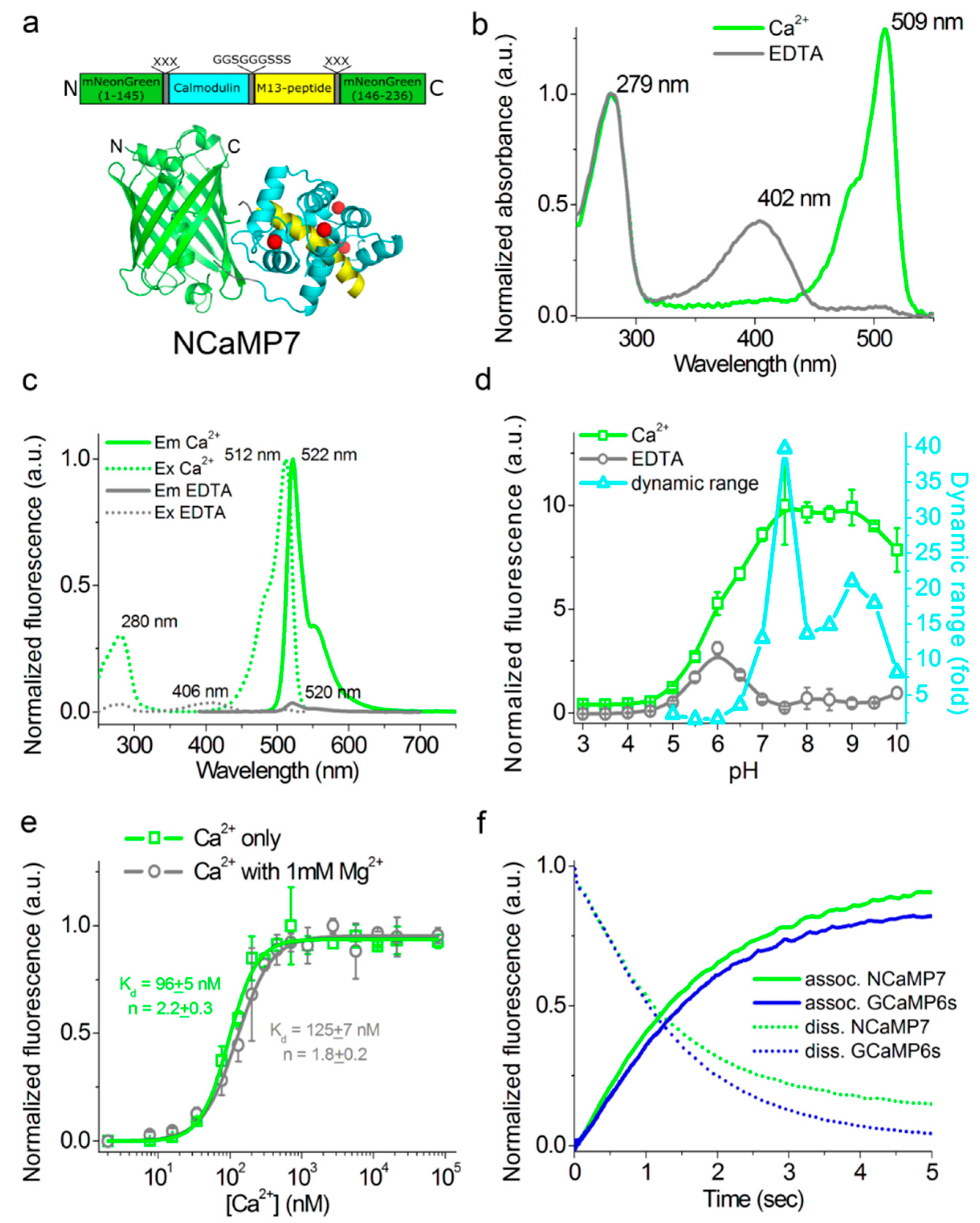
2.3. In Vitro Characterization of the Purified NCaMP Indicators
2.4. Characterization of the Kinetics for the NCaMP Calcium Indicators Using Stopped-Flow Fluorimetry
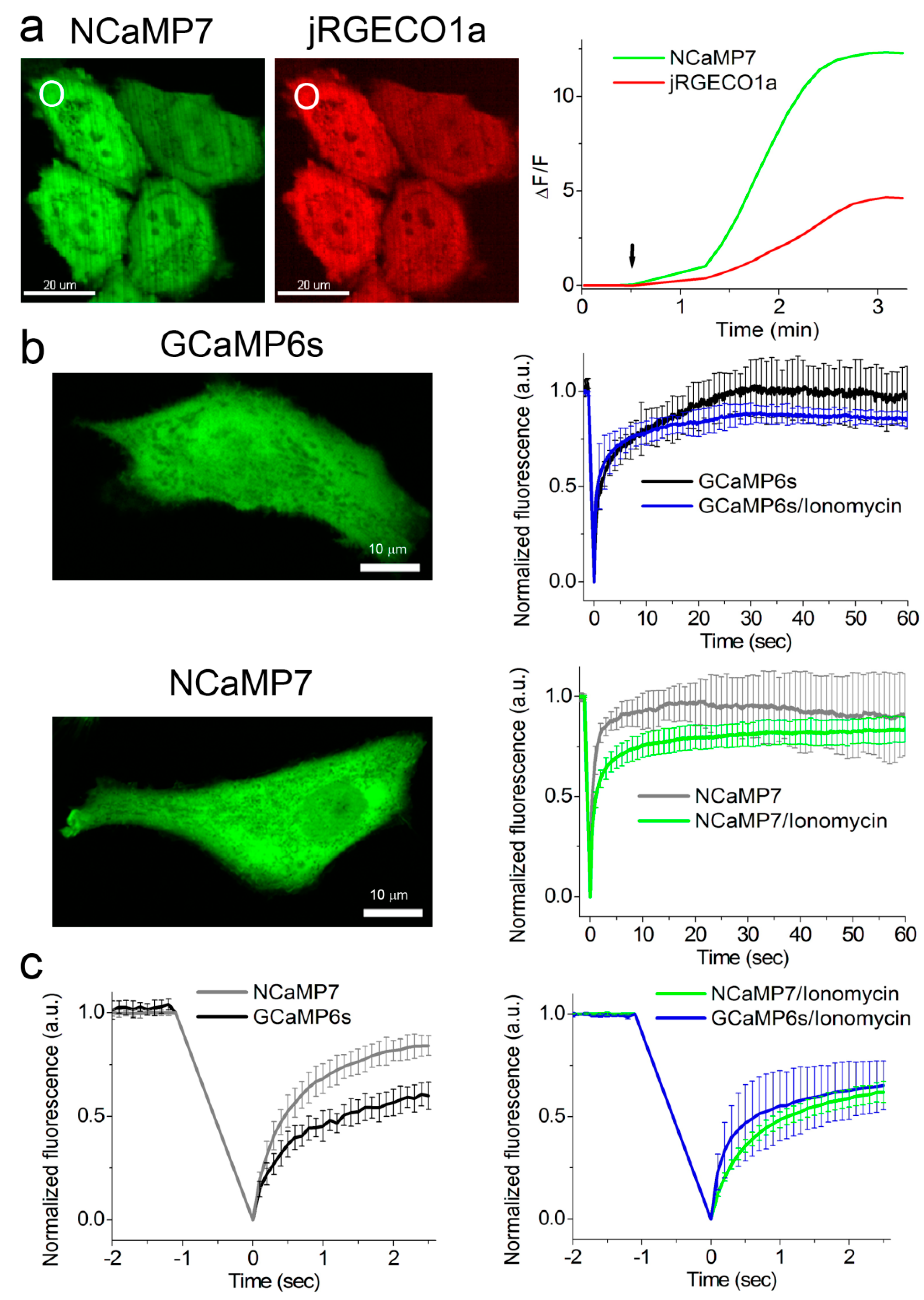
2.5. Calcium-Dependent Response of the NCaMP Calcium Indicators in HeLa Mammalian Cells
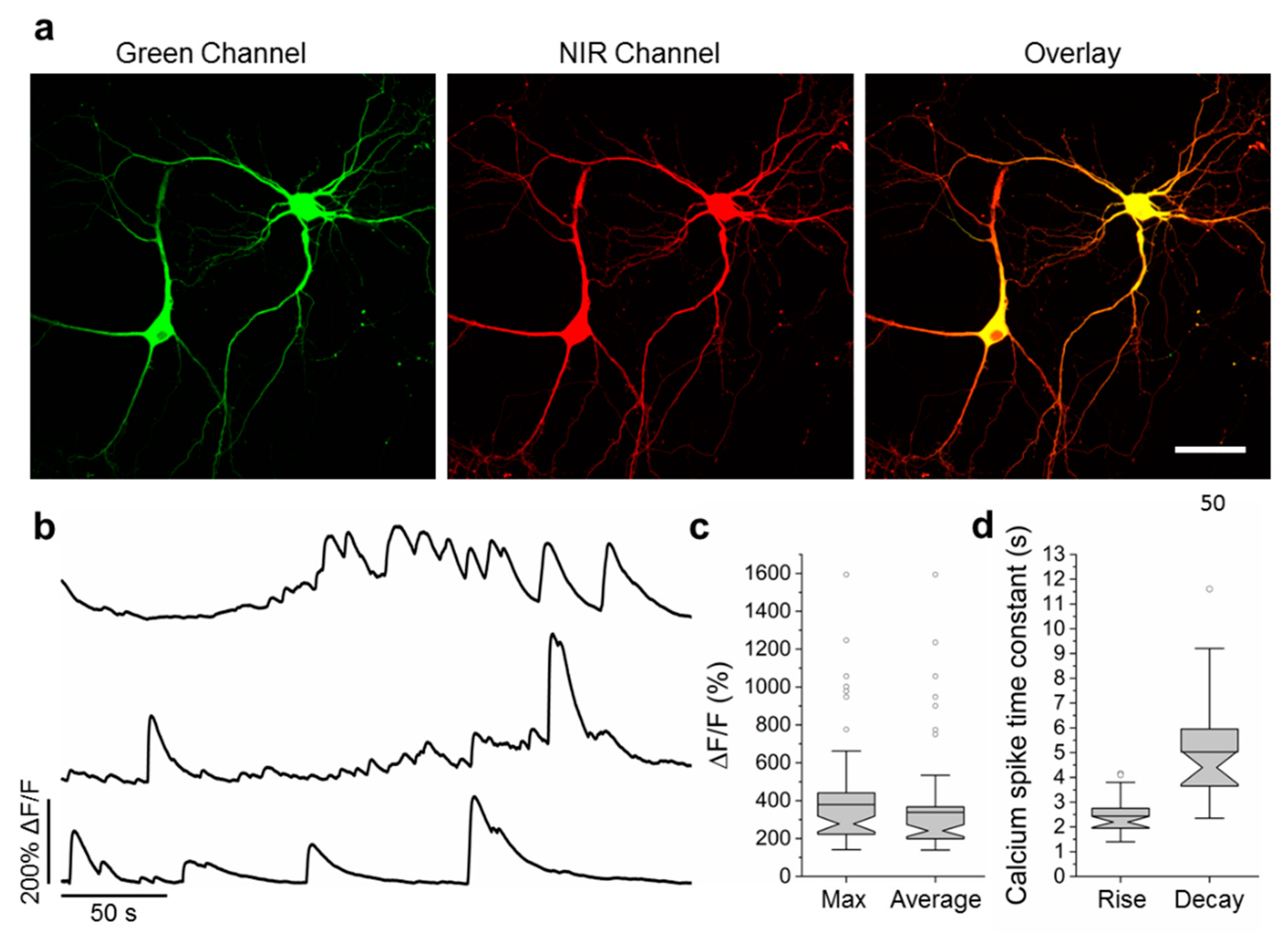
2.6. Visualization of Spontaneous and Induced Neuronal Activity in Dissociated Culture Using NCaMP Indicators and Confocal Imaging
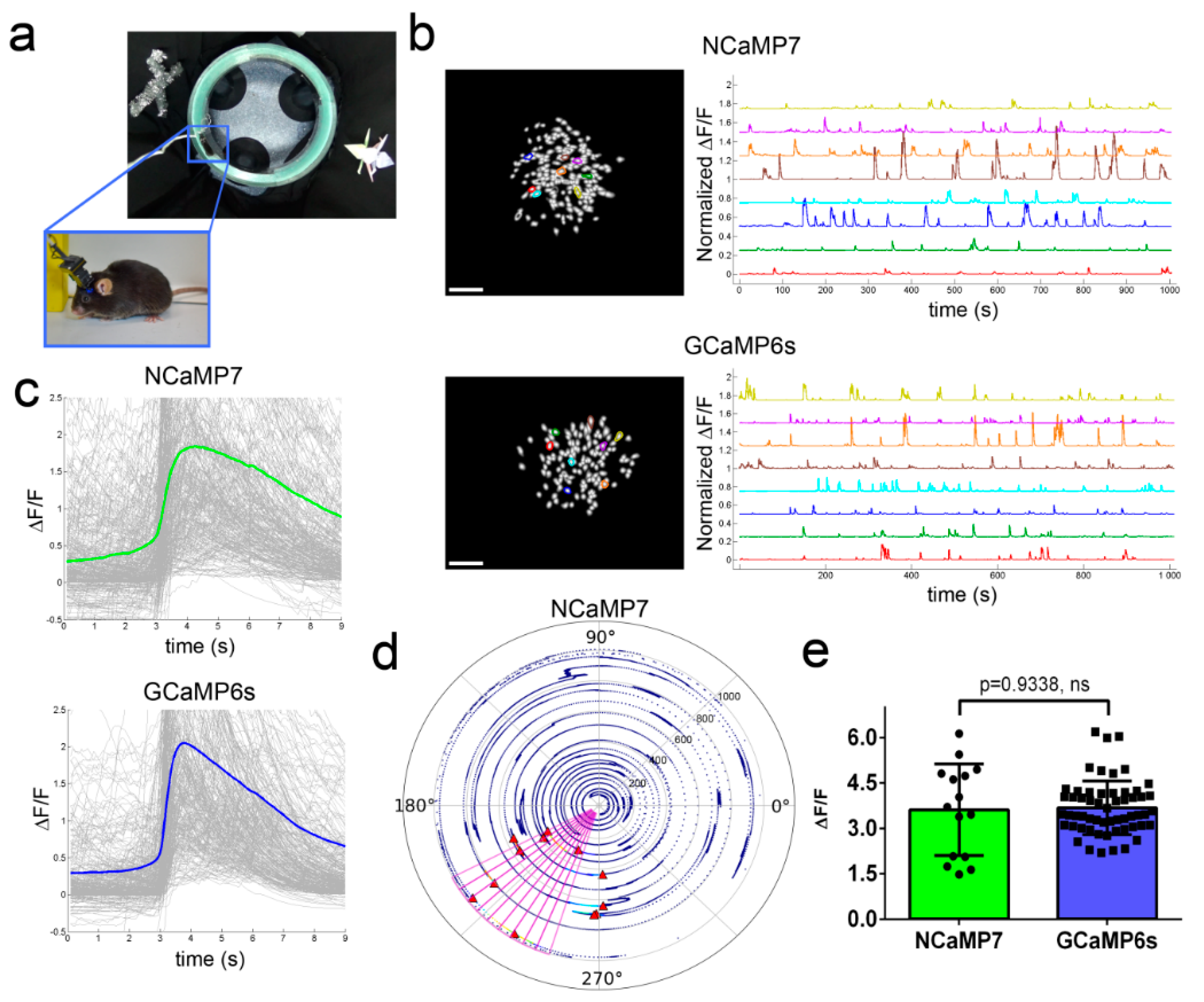
2.7. In Vivo Imaging of Neuronal Activity in the Hippocampus of Freely Moving Mice Using NCaMPs and an nVista Miniscope
2.8. In Vivo Two-Photon Imaging of Neuronal Activity in the Visual Cortex of Awake Mice Using the NCaMP7 Indicator
3. Materials and Methods
3.1. Mutagenesis and Library Screening
3.2. Proteins Purification and Characterization
3.3. Protein Purification for X-Ray Crystallography
3.4. Crystallization of NCaMP7
3.5. Data Collection, Processing, Structure Solution, and Refinement
3.6. Structure Analysis and Validation
3.7. Stopped-Flow Fluorimetry
3.8. Mammalian Plasmid Construction
3.9. Mammalian Live-Cell Imaging
3.10. Imaging in Primary Mouse Neuronal Cultures
3.11. Surgery and Imaging in Hippocampus Using an nVista HD Miniscope
3.12. Viral Injection to the Neonatal Mouse Brain and Surgery for in Vivo Two-Photon Imaging
3.13. Two-Photon Imaging in V1
3.14. Statistics
3.15. Ethical Approval and Animal Care
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| GECI | Genetically Encoded Calcium Indicator |
| EGFP | Enhanced green fluorescent protein |
| FP | Fluorescent Protein |
| AP | Action Potential |
| FRAP | Fluorescence Recovery After Photobleaching |
| H-bonds | Hydrogen bonds |
| DIVs | Days In Vitro |
| MIT | Massachusetts Institute of Technology |
| PDB ID | Protein Data Bank IDentifier |
| RMSD | Root-Mean-Square Deviation |
| TEV | Tobacco Etch Virus |
| TLS | Translation Libration Screw-motion refinement |
| QY | Quantum Yield |
| MAD | Median Absolute Deviation |
References
- Rodriguez, E.A.; Campbell, R.E.; Lin, J.Y.; Lin, M.Z.; Miyawaki, A.; Palmer, A.E.; Shu, X.; Zhang, J.; Tsien, R.Y. The Growing and Glowing Toolbox of Fluorescent and Photoactive proteins. Trends Biochem. Sci. 2017, 42, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Piatkevich, K.D.; Murdock, M.H.; Subach, F.V. Advances in engineering and application of optogenetic indicators for neuroscience. Appl. Sci. 2019, 9, 562. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Huang, C.; Li, J.Z.; Grewe, B.F.; Zhang, Y.; Eismann, S.; Schnitzer, M.J. High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 2015, 350, 1361–1366. [Google Scholar] [CrossRef]
- Tewson, P.H.; Martinka, S.; Shaner, N.C.; Hughes, T.E.; Quinn, A.M. New DAG and cAMP sensors optimized for live-cell assays in automated laboratories. J. Biomol. Screen. 2016, 21, 298–305. [Google Scholar] [CrossRef]
- Den Hamer, A.; Dierickx, P.; Arts, R.; de Vries, J.; Brunsveld, L.; Merkx, M. Bright bioluminescent BRET sensor proteins for measuring intracellular caspase activity. ACS Sens. 2017, 2, 729–734. [Google Scholar] [CrossRef]
- Subach, O.M.; Kunitsyna, T.A.; Mineyeva, O.A.; Lazutkin, A.A.; Bezryadnov, D.V.; Barykina, N.V.; Piatkevich, K.D.; Ermakova, Y.G.; Bilan, D.S.; Belousov, V.V.; et al. Slowly reducible genetically encoded green fluorescent indicator for In Vivo and Ex Vivo visualization of hydrogen peroxide. Int. J. Mol. Sci. 2019, 20, 3138. [Google Scholar] [CrossRef]
- Suzuki, K.; Kimura, T.; Shinoda, H.; Bai, G.; Daniels, M.J.; Arai, Y.; Nakano, M.; Nagai, T. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 2016, 7, 13718. [Google Scholar] [CrossRef]
- Barykina, N.V.; Subach, O.M.; Doronin, D.A.; Sotskov, V.P.; Roshchina, M.A.; Kunitsyna, T.A.; Malyshev, A.Y.; Smirnov, I.V.; Azieva, A.M.; Sokolov, I.S.; et al. A new design for a green calcium indicator with a smaller size and a reduced number of calcium-binding sites. Sci. Rep. 2016, 6, 34447. [Google Scholar] [CrossRef]
- Barykina, N.V.; Doronin, D.A.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Gruzdeva, A.M.; Kunitsyna, T.A.; Ivashkina, O.I.; Lazutkin, A.A.; et al. NTnC-like genetically encoded calcium indicator with a positive and enhanced response and fast kinetics. Sci. Rep. 2018, 8, 15233. [Google Scholar] [CrossRef]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar] [CrossRef] [PubMed]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Niino, Y. Molecular spies for bioimaging--fluorescent protein-based probes. Mol. Cell. 2015, 58, 632–643. [Google Scholar] [CrossRef]
- Doronin, D.A.; Barykina, N.V.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Isaakova, E.A.; Varizhuk, A.M.; Pozmogova, G.E.; Malyshev, A.Y.; et al. Genetically encoded calcium indicator with NTnC-like design and enhanced fluorescence contrast and kinetics. BMC Biotechnol. 2018, 18, 10. [Google Scholar] [CrossRef]
- Qian, Y.; Piatkevich, K.D.; Mc Larney, B.; Abdelfattah, A.S.; Mehta, S.; Murdock, M.H.; Gottschalk, S.; Molina, R.S.; Zhang, W.; Chen, Y.; et al. A genetically encoded near-infrared fluorescent calcium ion indicator. Nat. Methods 2019, 16, 171–174. [Google Scholar] [CrossRef]
- Subach, O.M.; Barykina, N.V.; Anokhin, K.V.; Piatkevich, K.D.; Subach, F.V. Near-Infrared Genetically Encoded Positive Calcium Indicator Based on GAF-FP Bacterial Phytochrome. Int J Mol Sci 2019, 20, 3488. [Google Scholar] [CrossRef]
- Qian, Y.; Rancic, V.; Wu, J.; Ballanyi, K.; Campbell, R.E. A bioluminescent Ca(2+) indicator based on a topological variant of GCaMP6s. Chembiochem Eur. J. Chem. Biol. 2018, 20, 516–520. [Google Scholar] [CrossRef]
- Moeyaert, B.; Holt, G.; Madangopal, R.; Perez-Alvarez, A.; Fearey, B.C.; Trojanowski, N.F.; Ledderose, J.; Zolnik, T.A.; Das, A.; Patel, D.; et al. Improved methods for marking active neuron populations. Nat. Commun. 2018, 9, 4440. [Google Scholar] [CrossRef]
- Subach, F.V.; Piatkevich, K.D.; Verkhusha, V.V. Directed molecular evolution to design advanced red fluorescent proteins. Nat. Methods 2011, 8, 1019–1026. [Google Scholar] [CrossRef]
- Ding, J.; Luo, A.F.; Hu, L.; Wang, D.; Shao, F. Structural basis of the ultrasensitive calcium indicator GCaMP6. Sci. China Life Sci. 2014, 57, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Clavel, D.; Gotthard, G.; von Stetten, D.; De Sanctis, D.; Pasquier, H.; Lambert, G.G.; Shaner, N.C.; Royant, A. Structural analysis of the bright monomeric yellow-green fluorescent protein mNeonGreen obtained by directed evolution. Acta Crystallogr. D Struct. Biol. 2016, 72, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Strynadka, N.C.; James, M.N. Crystal structures of the helix-loop-helix calcium-binding proteins. Annu. Rev. Biochem. 1989, 58, 951–998. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Barykina, N.V.; Subach, O.M.; Piatkevich, K.D.; Jung, E.E.; Malyshev, A.Y.; Smirnov, I.V.; Bogorodskiy, A.O.; Borshchevskiy, V.I.; Varizhuk, A.M.; Pozmogova, G.E.; et al. Green fluorescent genetically encoded calcium indicator based on calmodulin/M13-peptide from fungi. PLoS ONE 2017, 12, e0183757. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Subach, O.M.; Cranfill, P.J.; Davidson, M.W.; Verkhusha, V.V. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PloS One 2011, 6, e28674. [Google Scholar] [CrossRef] [PubMed]
- Grabe, M.; Oster, G. Regulation of organelle acidity. J. Gen. Physiol. 2001, 117, 329–344. [Google Scholar] [CrossRef]
- Brocard, J.B.; Rajdev, S.; Reynolds, I.J. Glutamate-induced increases in intracellular free Mg2+ in cultured cortical neurons. Neuron 1993, 11, 751–757. [Google Scholar] [CrossRef]
- Perez Koldenkova, V.; Nagai, T. Genetically encoded Ca(2+) indicators: Properties and evaluation. Biochim. Biophys. Acta 2013, 1833, 1787–1797. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Maravall, M.; Mainen, Z.F.; Sabatini, B.L.; Svoboda, K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 2000, 78, 2655–2667. [Google Scholar] [CrossRef]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 2016, 5. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, N.; He, Y.; Liu, Y.; Ge, L.; Zou, L.; Song, S.; Xiong, W.; Liu, X. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat. Commun. 2018, 9, 1504. [Google Scholar] [CrossRef]
- Piatkevich, K.D.; Jung, E.E.; Straub, C.; Linghu, C.; Park, D.; Suk, H.J.; Hochbaum, D.R.; Goodwin, D.; Pnevmatikakis, E.; Pak, N.; et al. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 2018, 14, 352–360. [Google Scholar] [CrossRef]
- Lu, J.; Li, C.; Singh-Alvarado, J.; Zhou, Z.C.; Frohlich, F.; Mooney, R.; Wang, F. MIN1PIPE: A miniscope 1-photon-based calcium imaging signal extraction pipeline. Cell Rep. 2018, 23, 3673–3684. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Padilla, J.E.; Yeates, T.O. A statistic for local intensity differences: Robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Pnevmatikakis, E.A.; Giovannucci, A. NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 2017, 291, 83–94. [Google Scholar] [CrossRef]
- Lopes, G.; Bonacchi, N.; Frazao, J.; Neto, J.P.; Atallah, B.V.; Soares, S.; Moreira, L.; Matias, S.; Itskov, P.M.; Correia, P.A.; et al. Bonsai: An event-based framework for processing and controlling data streams. Front. Neuroinform. 2015, 9, 7. [Google Scholar] [CrossRef]
- Kim, J.Y.; Grunke, S.D.; Levites, Y.; Golde, T.E.; Jankowsky, J.L. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J. Vis. Exp. 2014, 91, e51863. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ash, R.T.; Ceballos-Diaz, C.; Levites, Y.; Golde, T.E.; Smirnakis, S.M.; Jankowsky, J.L. Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits In Vivo. Eur. J. Neurosci. 2013, 37, 1203–1220. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, A.; Friedrich, J.; Gunn, P.; Kalfon, J.; Brown, B.L.; Koay, S.A.; Taxidis, J.; Najafi, F.; Gauthier, J.L.; Zhou, P.; et al. CaImAn an open source tool for scalable calcium imaging data analysis. Elife 2019, 8, e38173. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Sun, Y.; Mohar, B.; Hulse, B.K.; Kerlin, A.M.; Hasseman, J.P.; Tsegaye, G.; Tsang, A.; Wong, A.; Patel, R.; et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 2019, 16, 649–657. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Proteins | ||||||
|---|---|---|---|---|---|---|---|
| NCaMP7 | GCaMP6s | ||||||
| apo | sat | apo | sat | ||||
| Abs/Exc maximum (nm) | 402/406 | 509/512 | 402/ND | 500/ND | |||
| Emission maximum (nm) | 520 | 522 | 518 | 515 | |||
| Quantum yield a | 0.048 ± 0.003 | 0.52 ± 0.03 | 0.11 ± 0.01 | 0.61 | |||
| ε (mM−1 cm−1) b | 46.6 ± 2.7 | 110.0 ± 7.3 | 33.3 ± 0.6 | 77 ± 3 | |||
| Brightness (%) c | 6.5 | 179 | 8.3 | 107 | |||
| ΔF/F (fold) | 0 mM Mg2+ | 89 ± 27 | 43 ± 6 | ||||
| 1 mM Mg2+ | 27 ± 3 | 46 ± 24 | |||||
| pKa | 5.43 ± 0.09 6.62 ± 0.09 | 6.18 ± 0.21 | 9.6 ± 0.3 | 6.16 ± 0.08 | |||
| Kd (nM) d | 0 mM Mg2+ | 96 ± 5 (n = 2.2 ± 0.3) | 144±3 (n = 3.5 ± 0.2) | ||||
| 1 mM Mg2+ | 125 ± 7 (n = 1.8 ± 0.2) | 227.3 ± 0.2 | |||||
| kobs (s−1) e | 0.54 ± 0.02 | 0.49 ± 0.05 | |||||
| koff (s−1) f | k1 (contrib., %) | 0.89 ± 0.01 (78 ± 1) | 0.69 ± 0.01 | ||||
| k2 (contrib., %) | 0.11 ± 0.01 (22 ± 1) | ||||||
| t1/2, s g | 1.1 ± 0.1 | 1.01 ± 0.04 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Gruzdeva, A.M.; Barykina, N.V.; Ivashkina, O.I.; Anokhin, K.V.; Nikolaeva, A.Y.; Korzhenevskiy, D.A.; Vlaskina, A.V.; et al. Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. Int. J. Mol. Sci. 2020, 21, 1644. https://doi.org/10.3390/ijms21051644
Subach OM, Sotskov VP, Plusnin VV, Gruzdeva AM, Barykina NV, Ivashkina OI, Anokhin KV, Nikolaeva AY, Korzhenevskiy DA, Vlaskina AV, et al. Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. International Journal of Molecular Sciences. 2020; 21(5):1644. https://doi.org/10.3390/ijms21051644
Chicago/Turabian StyleSubach, Oksana M., Vladimir P. Sotskov, Viktor V. Plusnin, Anna M. Gruzdeva, Natalia V. Barykina, Olga I. Ivashkina, Konstantin V. Anokhin, Alena Y. Nikolaeva, Dmitry A. Korzhenevskiy, Anna V. Vlaskina, and et al. 2020. "Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein" International Journal of Molecular Sciences 21, no. 5: 1644. https://doi.org/10.3390/ijms21051644
APA StyleSubach, O. M., Sotskov, V. P., Plusnin, V. V., Gruzdeva, A. M., Barykina, N. V., Ivashkina, O. I., Anokhin, K. V., Nikolaeva, A. Y., Korzhenevskiy, D. A., Vlaskina, A. V., Lazarenko, V. A., Boyko, K. M., Rakitina, T. V., Varizhuk, A. M., Pozmogova, G. E., Podgorny, O. V., Piatkevich, K. D., Boyden, E. S., & Subach, F. V. (2020). Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. International Journal of Molecular Sciences, 21(5), 1644. https://doi.org/10.3390/ijms21051644