Upregulation of Thr/Tyr kinase Increases the Cancer Progression by Neurotensin and Dihydropyrimidinase-Like 3 in Lung Cancer

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Elevated TTK Expression Conferred Poor Prognosis in Lung Cancer Patients
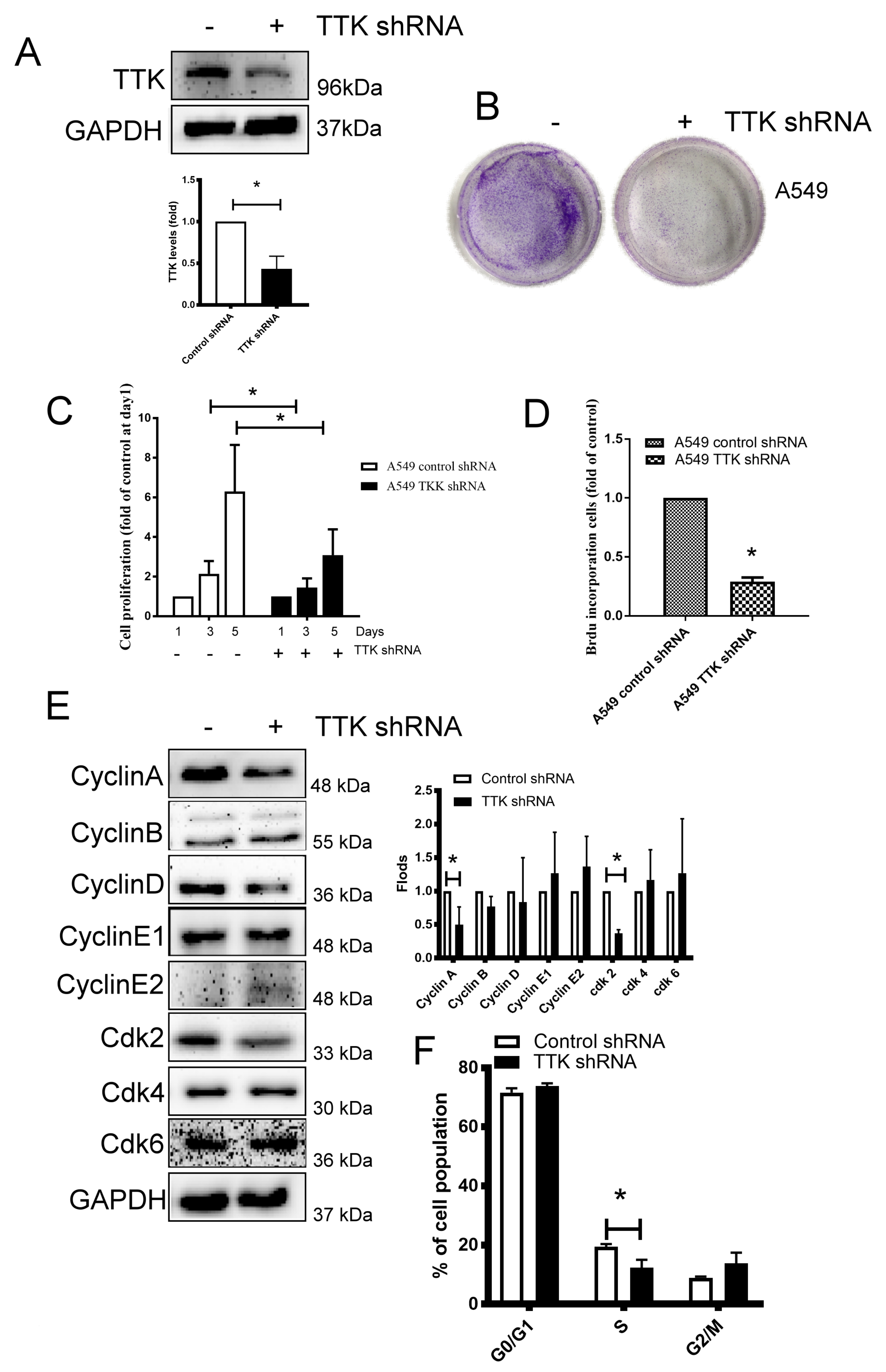
2.2. TTK Decreased Cancer Growth by Interfering Cell Cycle Progression in Lung Cancer
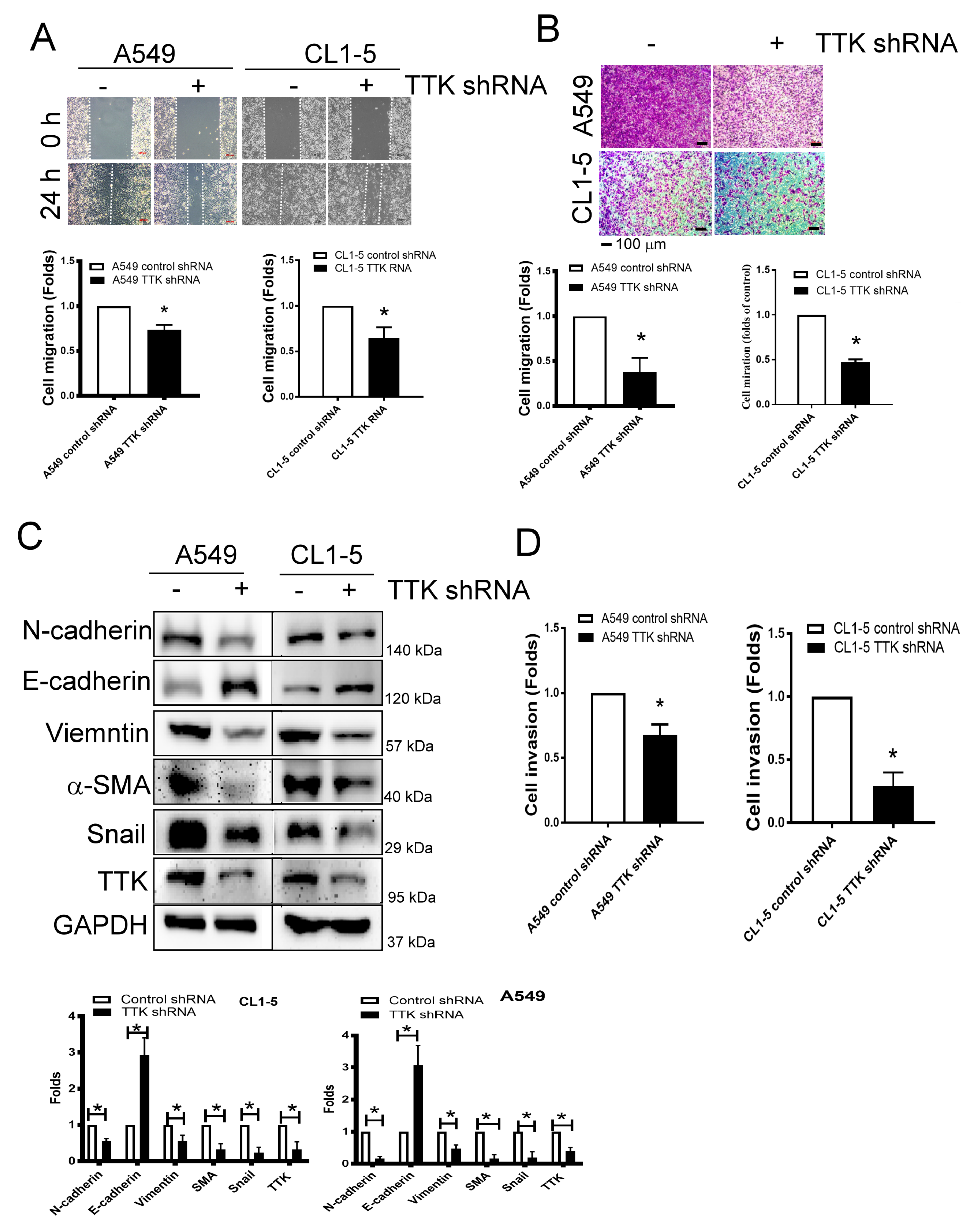
2.3. TTK Regulated Metastatic Behaviors and EMT in Lung Cancer
2.4. NTS and DYPSL3 Act as Downstream Regulators of TTK
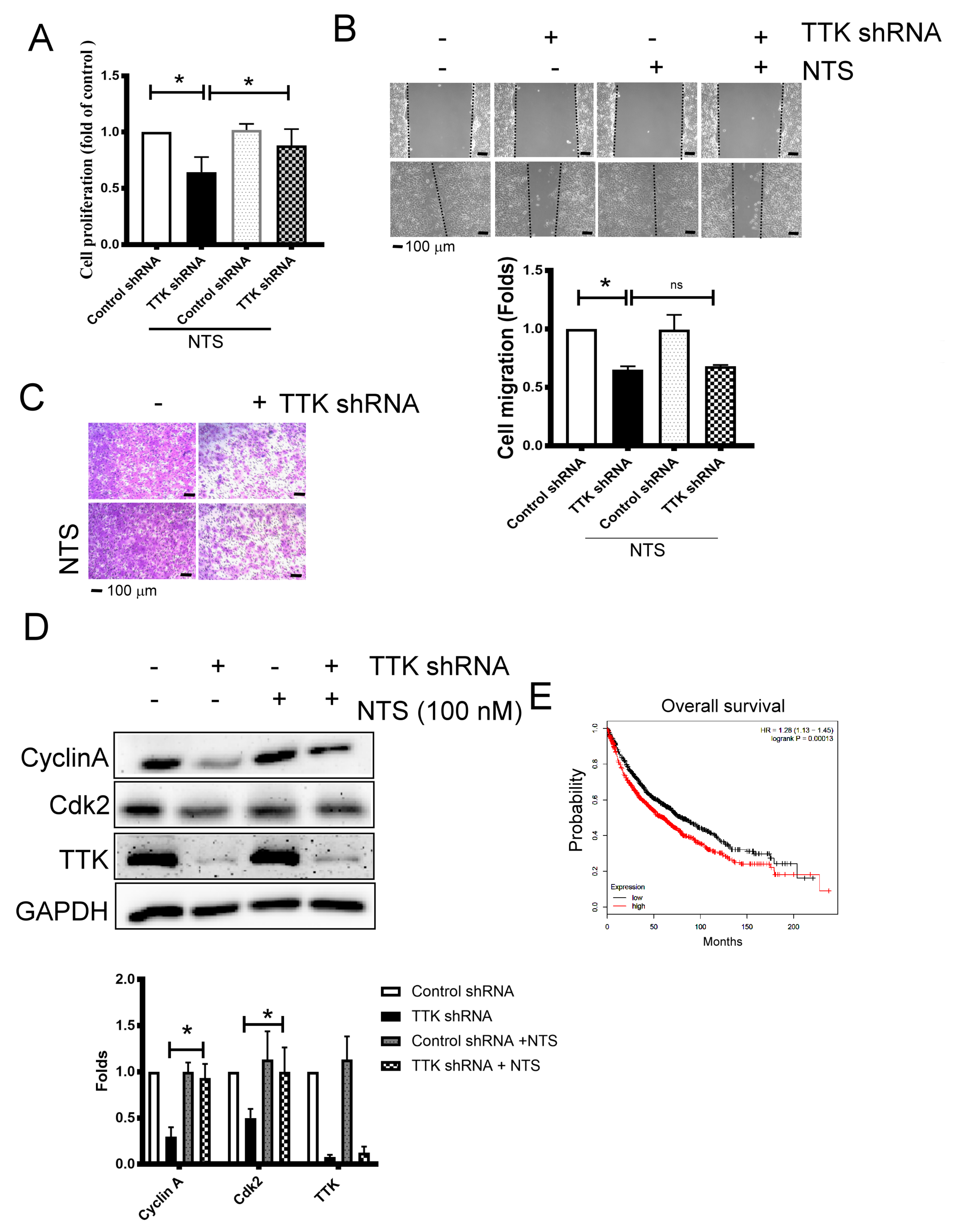
2.5. NTS Is Involved in TTK- Related Cell Proliferation Regulation
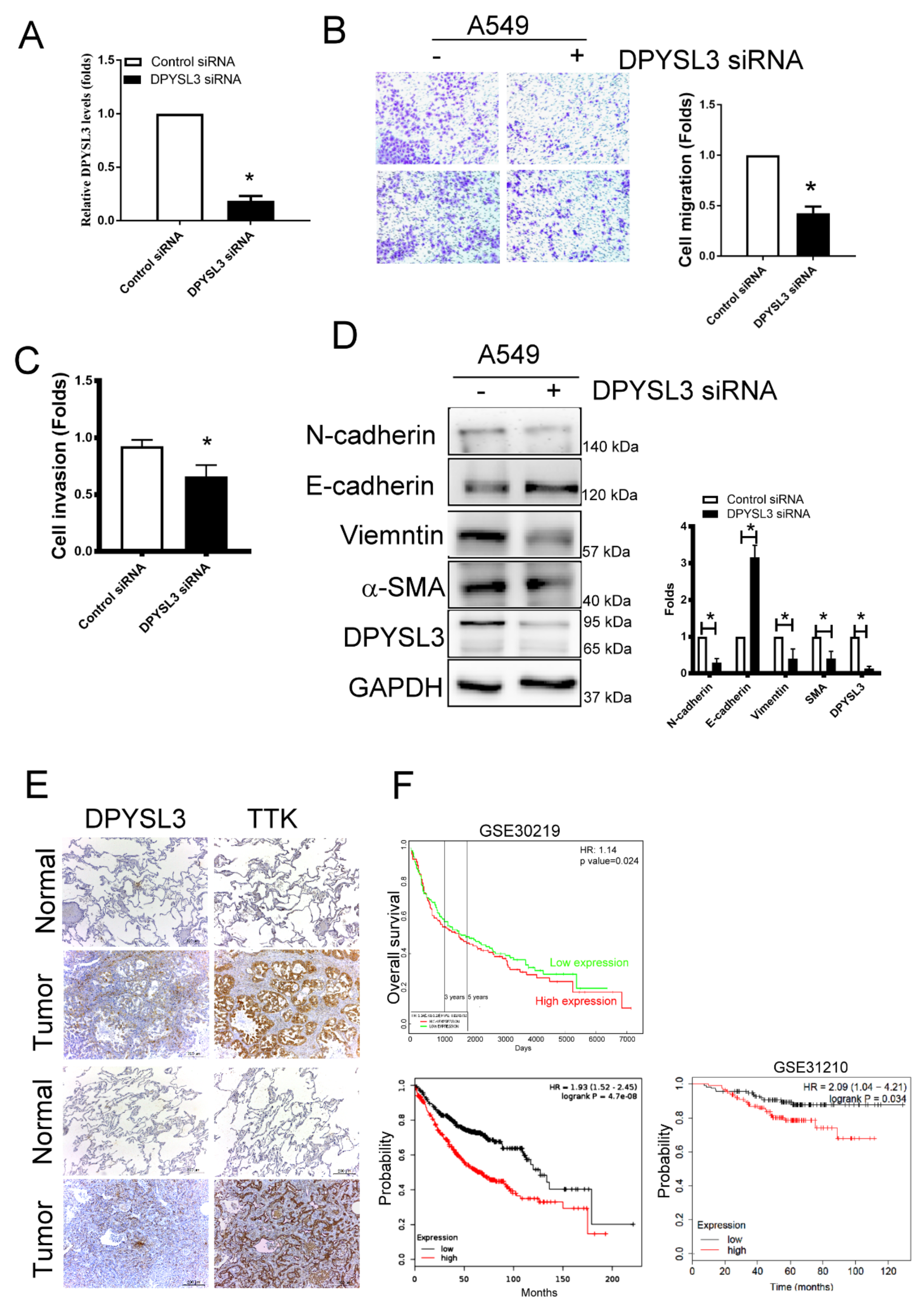
2.6. DPYSL3 Contributes the Enhancement of Cell Migration and EMT
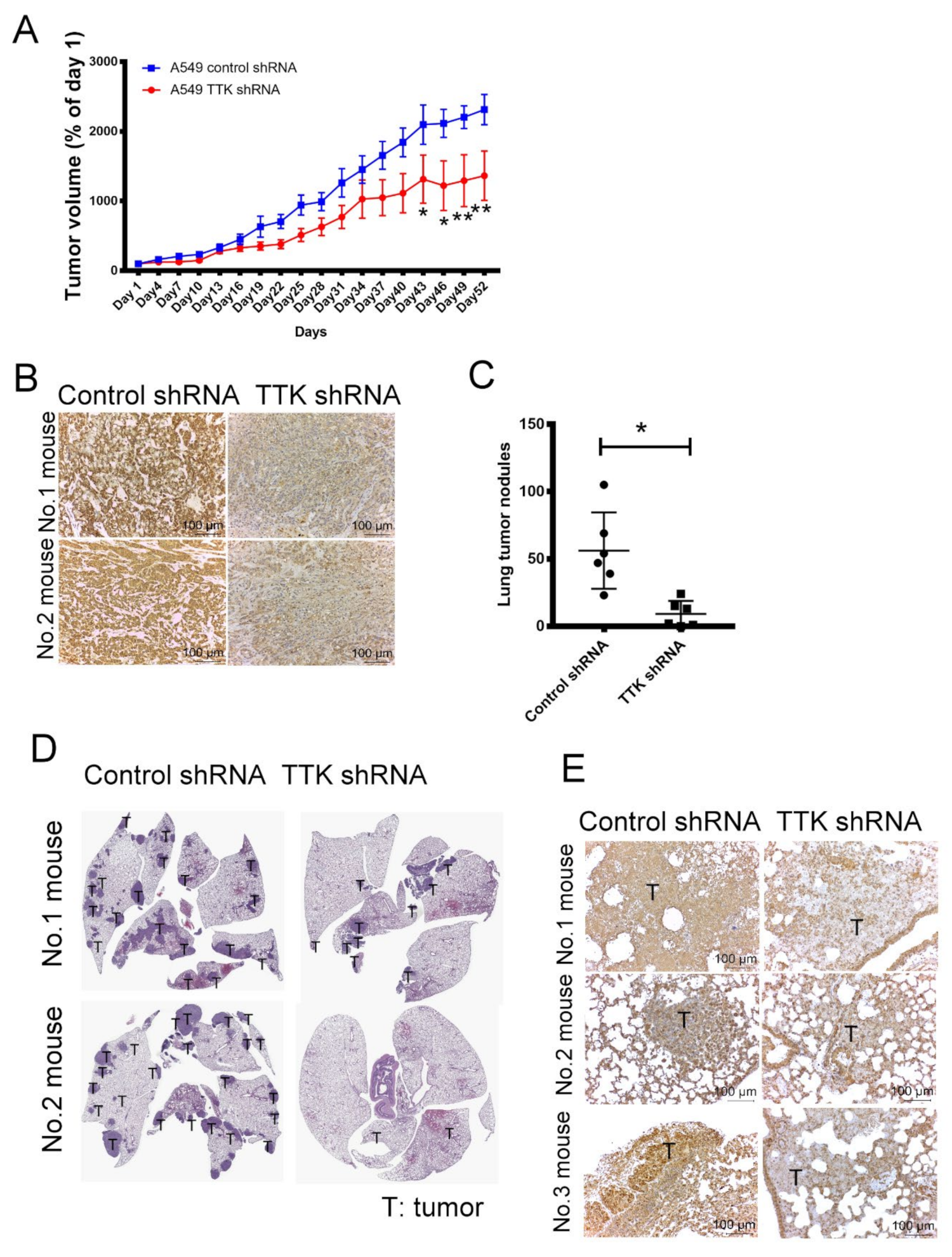
2.7. Inhibition of TTK Suppressed Cancer Growth and Lung Metastasis In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Bioinformatics
4.3. NGS and Microarray
4.4. RNA-Sequencing and Quantitative Real-Time Polymerase Chain Reaction (Qrt-PCR)
4.5. Immunoblot and NTS Determination
4.6. TTK and DPYSL3 Knockdown
4.7. Cell Proliferation, Colony Formation and 5-Bromo-2-Deoxyuridine (Brdu) Incorporation
4.8. Cell Migration and Invasion
4.9. Cell Cycle Analysis
4.10. Animal Models
4.11. Immunohistochemistry (IHC)
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DPYSL3 | dihydropyrimidinase-like 3 |
| EMT | epithelial–mesenchymal transition |
| Mps1 | monopolar spindle 1 |
| NGS | next-generation sequencing |
| NTS | neurotensin |
| SAC | spindle assembly checkpoint |
| TTK | Thr/Tyr kinase |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Wakai, T.; Prasoon, P.; Hirose, Y.; Shimada, Y.; Ichikawa, H.; Nagahashi, M. Next-generation sequencing-based clinical sequencing: toward precision medicine in solid tumors. Int. J. Clin. 2019, 24, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef]
- Stratford, J.K.; Yan, F.; Hill, R.A.; Major, M.B.; Graves, L.M.; Der, C.J.; Yeh, J.J. Genetic and pharmacological inhibition of TTK impairs pancreatic cancer cell line growth by inducing lethal chromosomal instability. PLoS ONE 2017, 12, e0174863. [Google Scholar] [CrossRef]
- Xu, Q.; Xu, Y.; Pan, B.; Wu, L.; Ren, X.; Zhou, Y.; Mao, F.; Lin, Y.; Guan, J.; Shen, S.; et al. TTK is a favorable prognostic biomarker for triple-negative breast cancer survival. Oncotarget 2016, 7, 81815–81829. [Google Scholar] [CrossRef]
- Zhu, D.; Xu, S.; Deyanat-Yazdi, G.; Peng, S.X.; Barnes, L.A.; Narla, R.K.; Tran, T.; Mikolon, D.; Ning, Y.; Shi, T.; et al. Synthetic Lethal Strategy Identifies a Potent and Selective TTK and CLK1/2 Inhibitor for Treatment of Triple-Negative Breast Cancer with a Compromised G1-S Checkpoint. Mol. Cancer Ther. 2018, 17, 1727–1738. [Google Scholar] [CrossRef]
- Miao, R.; Wu, Y.; Zhang, H.; Zhou, H.; Sun, X.; Csizmadia, E.; He, L.; Zhao, Y.; Jiang, C.; Miksad, R.A.; et al. Utility of the dual-specificity protein kinase TTK as a therapeutic target for intrahepatic spread of liver cancer. Sci. Rep. 2016, 6, 33121. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Gao, J.; Zhu, H.; Cui, B.; Zhang, T.; Zhou, Y.; Liu, Q.; He, H.; Xiao, R.; et al. A novel USP9X substrate TTK contributes to tumorigenesis in non-small-cell lung cancer. Theranostics 2018, 8, 2348–2360. [Google Scholar] [CrossRef]
- Thu, K.L.; Silvester, J.; Elliott, M.J.; Ba-Alawi, W.; Duncan, M.H.; Elia, A.C.; Mer, A.S.; Smirnov, P.; Safikhani, Z.; Haibe-Kains, B.; et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E1570–e1577. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Lee, Y.L.; Chen, F.W.; Chang, K.F.; Chang, W.A.; Tsai, Y.M.; Chong, I.W.; Kuo, P.L. Identification of novel gene expression signature in lung adenocarcinoma by using next-generation sequencing data and bioinformatics analysis. Oncotarget 2017, 8, 104831–104854. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, J.A. Increased serum level of RPMPS-1/S27 protein in patients with various types of cancer is useful for the early detection, prevention and therapy. Cancer Genom. Proteom. 2012, 9, 203–256. [Google Scholar]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–w102. [Google Scholar] [CrossRef]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef]
- Hiruma, Y.; Sacristan, C.; Pachis, S.T.; Adamopoulos, A.; Kuijt, T.; Ubbink, M.; von Castelmur, E.; Perrakis, A.; Kops, G.J. CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. N. Y. Sci. J. 2015, 348, 1264–1267. [Google Scholar] [CrossRef]
- Van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef]
- Liu, X.; Liao, W.; Yuan, Q.; Ou, Y.; Huang, J. TTK activates Akt and promotes proliferation and migration of hepatocellular carcinoma cells. Oncotarget 2015, 6, 34309–34320. [Google Scholar] [CrossRef]
- Kaistha, B.P.; Honstein, T.; Muller, V.; Bielak, S.; Sauer, M.; Kreider, R.; Fassan, M.; Scarpa, A.; Schmees, C.; Volkmer, H.; et al. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br. J. Cancer 2014, 111, 1780–1787. [Google Scholar] [CrossRef]
- Abbaci, A.; Talbot, H.; Saada, S.; Gachard, N.; Abraham, J.; Jaccard, A.; Bordessoule, D.; Fauchais, A.L.; Naves, T.; Jauberteau, M.O. Neurotensin receptor type 2 protects B-cell chronic lymphocytic leukemia cells from apoptosis. Oncogene 2018, 37, 756–767. [Google Scholar] [CrossRef]
- Zhu, S.; Tian, H.; Niu, X.; Wang, J.; Li, X.; Jiang, N.; Wen, S.; Chen, X.; Ren, S.; Xu, C.; et al. Neurotensin and its receptors mediate neuroendocrine transdifferentiation in prostate cancer. Oncogene 2019, 38, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.J.; Liu, X.Y.; Zhang, J.H.; Ke, S.Y.; Fei, H.J. Neurotensin promotes cholangiocarcinoma metastasis via the EGFR/AKT pathway. Gene 2019, 687, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Long, X.; Zhang, L.; Ye, Y.; Guo, J.; Liu, P.; Zhang, R.; Ning, J.; Yu, W.; Wei, F.; et al. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology 2018, 7, e1440166. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Zhou, J.; Yang, W.; Cui, H.; Xu, M.; Yi, L. Oncogenic role of neurotensin and neurotensin receptors in various cancers. Clin. Exp. Pharmacol. Physiol. 2017, 44, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Dobner, P.R.; Carraway, R.E. Involvement of MAP-kinase, PI3-kinase and EGF-receptor in the stimulatory effect of Neurotensin on DNA synthesis in PC3 cells. Regul. Pept. 2004, 120, 155–166. [Google Scholar] [CrossRef]
- Massa, F.; Tormo, A.; Beraud-Dufour, S.; Coppola, T.; Mazella, J. Neurotensin-induced Erk1/2 phosphorylation and growth of human colonic cancer cells are independent from growth factors receptors activation. Biochem. Biophys. Res. Commun. 2011, 414, 118–122. [Google Scholar] [CrossRef]
- Guha, S.; Lunn, J.A.; Santiskulvong, C.; Rozengurt, E. Neurotensin stimulates protein kinase C-dependent mitogenic signaling in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2003, 63, 2379–2387. [Google Scholar]
- Ocejo-Garcia, M.; Ahmed, S.I.; Coulson, J.M.; Woll, P.J. Use of RT-PCR to detect co-expression of neuropeptides and their receptors in lung cancer. Lung Cancer 2001, 33, 1–9. [Google Scholar] [CrossRef]
- Tan, F.; Wahdan-Alaswad, R.; Yan, S.; Thiele, C.J.; Li, Z. Dihydropyrimidinase-like protein 3 expression is negatively regulated by MYCN and associated with clinical outcome in neuroblastoma. Cancer Sci. 2013, 104, 1586–1592. [Google Scholar] [CrossRef]
- Manivannan, J.; Tay, S.S.; Ling, E.A.; Dheen, S.T. Dihydropyrimidinase-like 3 regulates the inflammatory response of activated microglia. Neuroscience 2013, 253, 40–54. [Google Scholar] [CrossRef]
- Li, B.; Li, C. Suppression of Prostate Cancer Metastasis by DPYSL3-Targeted saRNA. Exp. Med. Biol. 2017, 983, 207–216. [Google Scholar] [CrossRef]
- Li, C.; Jiang, W.; Hu, Q.; Li, L.C.; Dong, L.; Chen, R.; Zhang, Y.; Tang, Y.; Thrasher, J.B.; Liu, C.B.; et al. Enhancing DPYSL3 gene expression via a promoter-targeted small activating RNA approach suppresses cancer cell motility and metastasis. Oncotarget 2016, 7, 22893–22910. [Google Scholar] [CrossRef] [PubMed]
- Oya, H.; Kanda, M.; Sugimoto, H.; Shimizu, D.; Takami, H.; Hibino, S.; Hashimoto, R.; Okamura, Y.; Yamada, S.; Fujii, T.; et al. Dihydropyrimidinase-like 3 is a putative hepatocellular carcinoma tumor suppressor. Gastroenterology 2015, 50, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Matsunuma, R.; Chan, D.W.; Kim, B.J.; Singh, P.; Han, A.; Saltzman, A.B.; Cheng, C.; Lei, J.T.; Wang, J.; Roberto da Silva, L.; et al. DPYSL3 modulates mitosis, migration, and epithelial-to-mesenchymal transition in claudin-low breast cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E11978–e11987. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Arai, T.; Sugawara, S.; Okato, A.; Kato, M.; Kojima, S.; Yamazaki, K.; Naya, Y.; Ichikawa, T.; Seki, N. Impact of novel oncogenic pathways regulated by antitumor miR-451a in renal cell carcinoma. Cancer Sci. 2018, 109, 1239–1253. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Peng, J.; Zhai, E.; He, Y.; Wu, H.; Chen, C.; Ma, J.; Wang, Z.; Cai, S. VEGF promotes gastric cancer development by upregulating CRMP4. Oncotarget 2016, 7, 17074–17086. [Google Scholar] [CrossRef]
- Kanda, M.; Nomoto, S.; Oya, H.; Shimizu, D.; Takami, H.; Hibino, S.; Hashimoto, R.; Kobayashi, D.; Tanaka, C.; Yamada, S.; et al. Dihydropyrimidinase-like 3 facilitates malignant behavior of gastric cancer. J. Exp. Clin. Cancer Res. 2014, 33, 66. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-M.; Wu, K.-L.; Chang, Y.-Y.; Hung, J.-Y.; Chang, W.-A.; Chang, C.-Y.; Jian, S.-F.; Tsai, P.-H.; Huang, Y.-C.; Chong, I.-W.; et al. Upregulation of Thr/Tyr kinase Increases the Cancer Progression by Neurotensin and Dihydropyrimidinase-Like 3 in Lung Cancer. Int. J. Mol. Sci. 2020, 21, 1640. https://doi.org/10.3390/ijms21051640
Tsai Y-M, Wu K-L, Chang Y-Y, Hung J-Y, Chang W-A, Chang C-Y, Jian S-F, Tsai P-H, Huang Y-C, Chong I-W, et al. Upregulation of Thr/Tyr kinase Increases the Cancer Progression by Neurotensin and Dihydropyrimidinase-Like 3 in Lung Cancer. International Journal of Molecular Sciences. 2020; 21(5):1640. https://doi.org/10.3390/ijms21051640
Chicago/Turabian StyleTsai, Ying-Ming, Kuan-Li Wu, Yung-Yun Chang, Jen-Yu Hung, Wei-An Chang, Chao-Yuan Chang, Shu-Fang Jian, Pei-Hsun Tsai, Yung-Chi Huang, Inn-Wen Chong, and et al. 2020. "Upregulation of Thr/Tyr kinase Increases the Cancer Progression by Neurotensin and Dihydropyrimidinase-Like 3 in Lung Cancer" International Journal of Molecular Sciences 21, no. 5: 1640. https://doi.org/10.3390/ijms21051640
APA StyleTsai, Y.-M., Wu, K.-L., Chang, Y.-Y., Hung, J.-Y., Chang, W.-A., Chang, C.-Y., Jian, S.-F., Tsai, P.-H., Huang, Y.-C., Chong, I.-W., & Hsu, Y.-L. (2020). Upregulation of Thr/Tyr kinase Increases the Cancer Progression by Neurotensin and Dihydropyrimidinase-Like 3 in Lung Cancer. International Journal of Molecular Sciences, 21(5), 1640. https://doi.org/10.3390/ijms21051640