Targeting Neurovascular Interaction in Retinal Disorders

 , ,
, ,  , and
, and 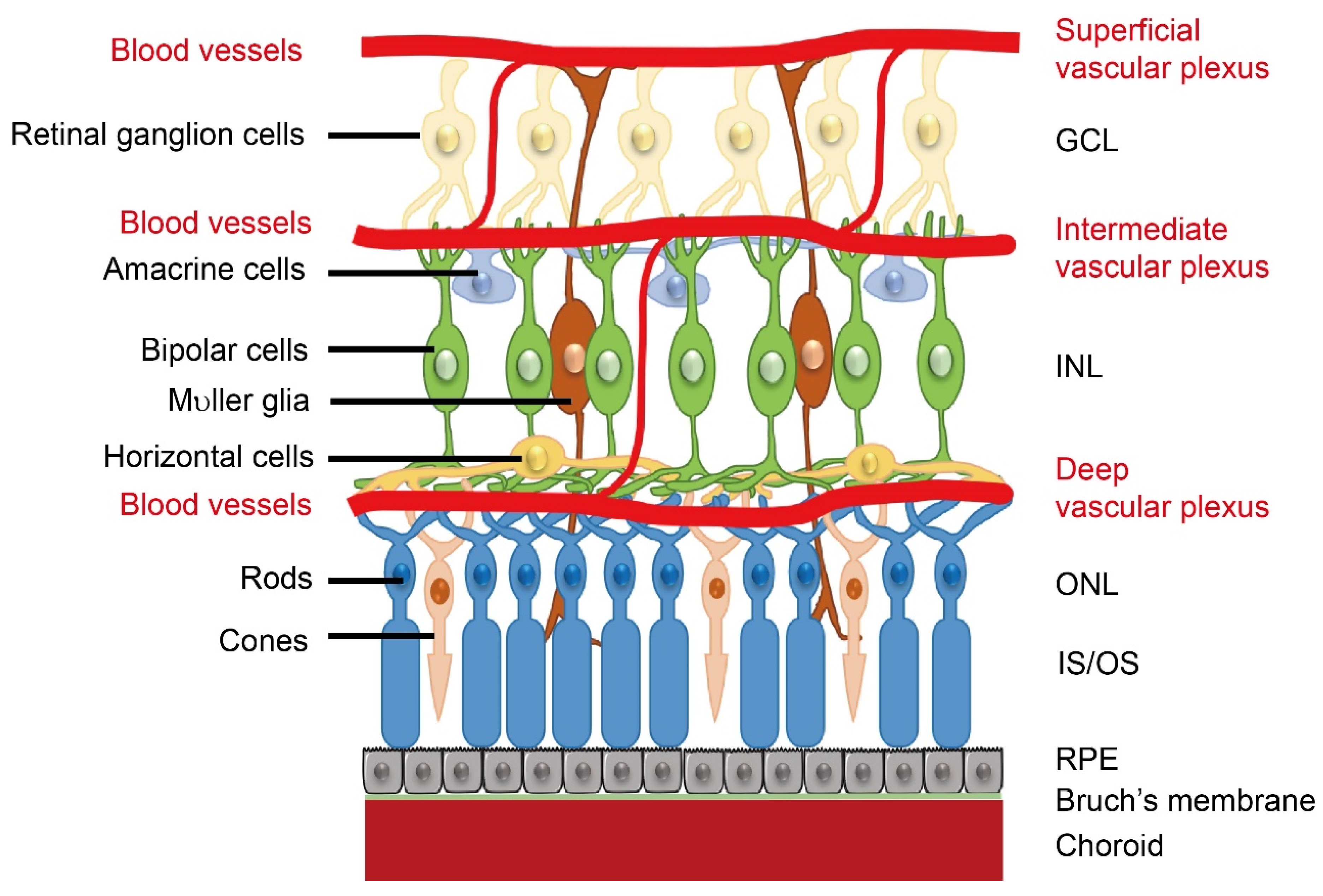{kind=link}
{kind=link}
{kind=link}
Abstract
1. Retinal Neurovascular Development
2. Factors Influence Retinal Neurovascular Interaction
2.1. Oxygen Shortage and Retinal Neovascularization
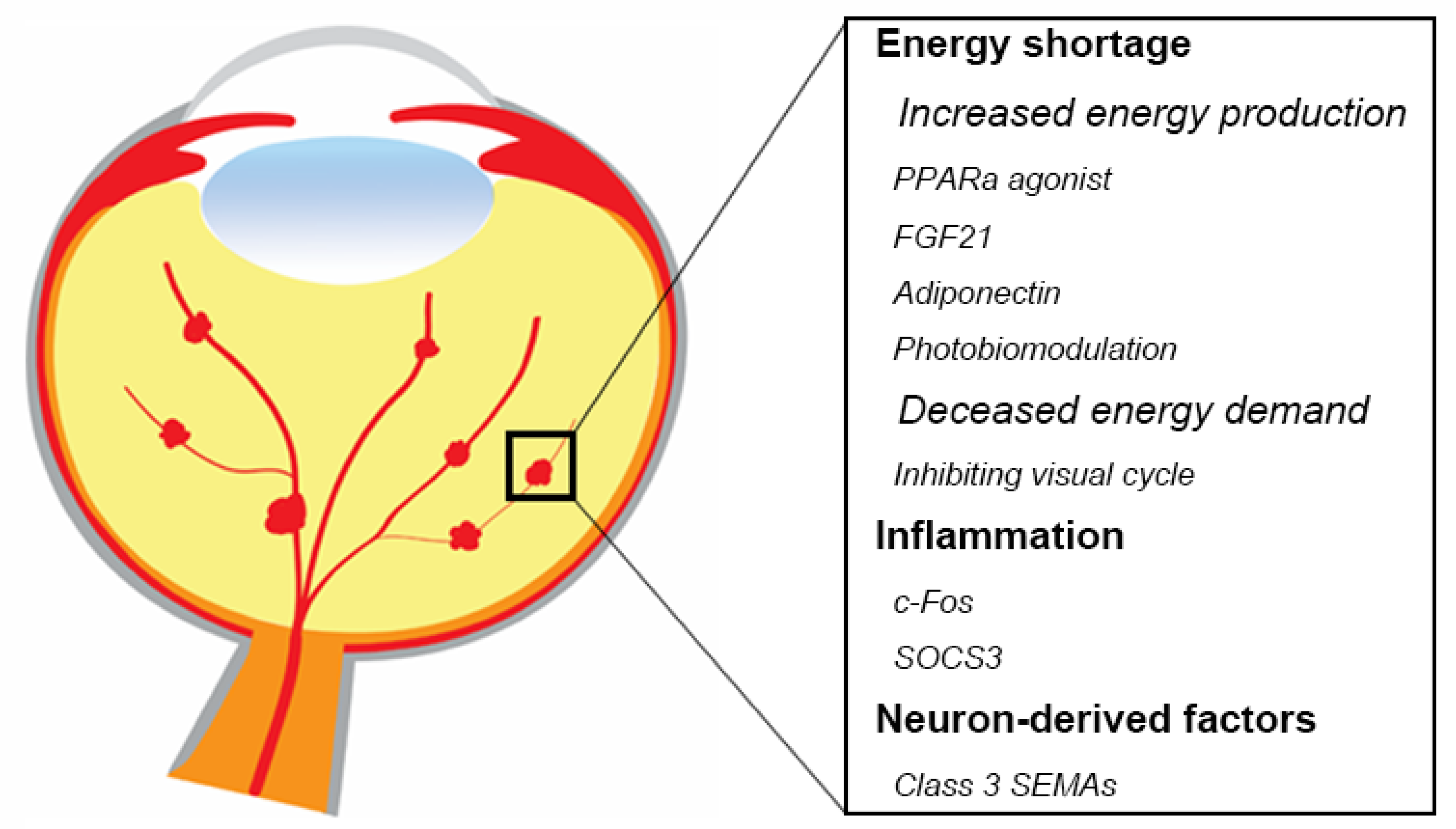
2.2. Energy Shortage Drives Retinal Neovascularization
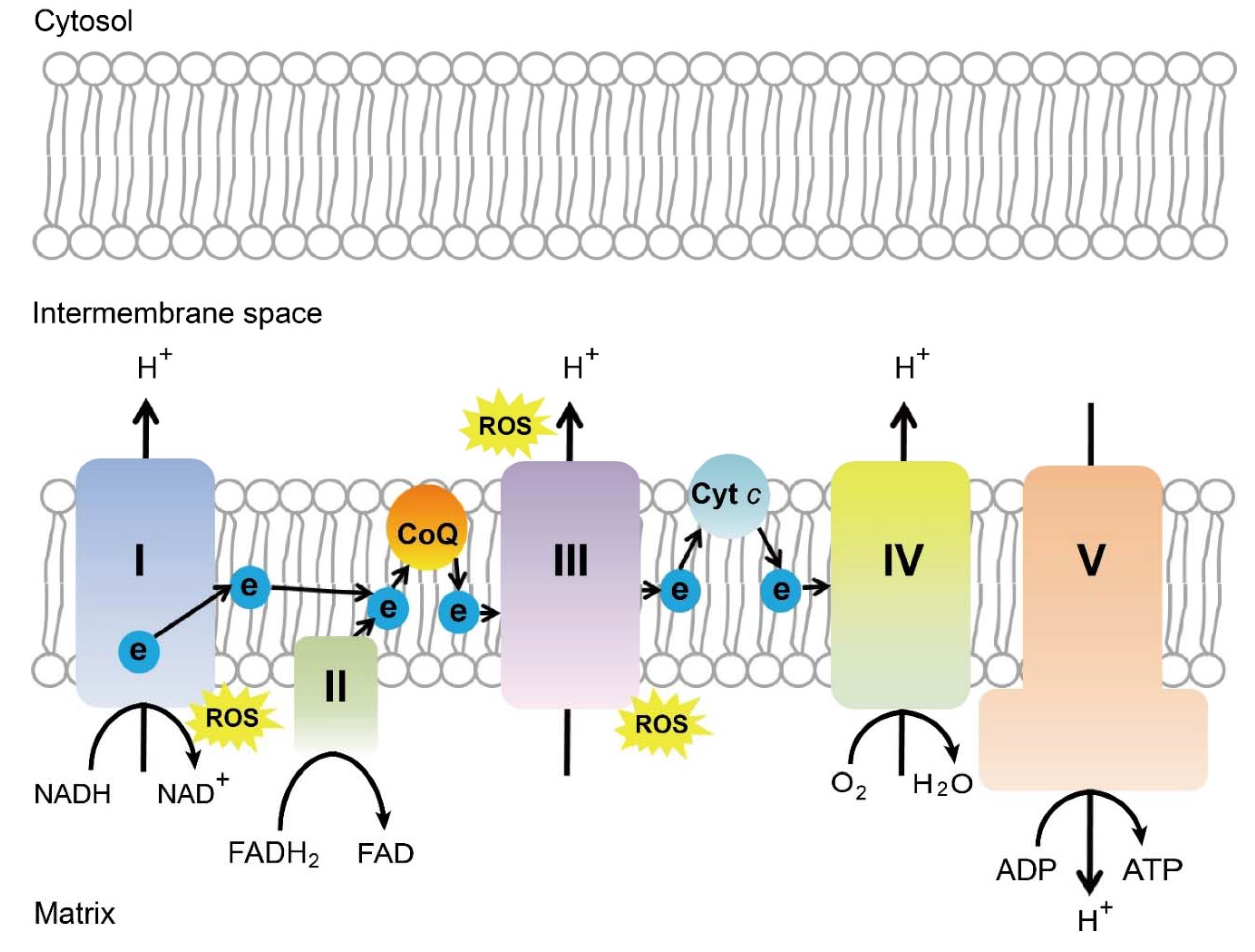
2.3. Oxidative Stress and Retinal Neovascularization
2.4. Retinal Circuitry and Retinal Remodeling
2.5. Inflammation and Retinal Neovascularization
2.6. Neuron-Derived Factors and Neovascularization
3. Therapeutic Potentials of Manipulating Pathways Controlling Neurovascular Crosstalk
3.1. Hormonal Modulation
3.2. Photobiomodulation
3.3. Suppression of the Visual Cycle to Decrease Energy Demands
3.4. Inflammatory Regulation
3.5. Class 3 SEMAs
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Oyster, C.W. The Human Eye: Structure and Function; Sinauer Associates: Sunderland, MA, USA, 1999. [Google Scholar]
- Hendrickson, A. A morphological comparison of foveal development in man and monkey. Eye (Lond.) 1992, 6, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; D’Amore, P.A. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int. J. Dev. Biol. 2004, 48, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, C.H.; Huang, S.; Chen, J. Wnt Signaling in vascular eye diseases. Prog. Retin. Eye Res. 2019, 70, 110–133. [Google Scholar] [CrossRef]
- Ye, X.; Wang, Y.; Cahill, H.; Yu, M.; Badea, T.C.; Smallwood, P.M.; Peachey, N.S.; Nathans, J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 2009, 139, 285–298. [Google Scholar] [CrossRef]
- Wang, Y.; Cho, C.; Williams, J.; Smallwood, P.M.; Zhang, C.; Junge, H.J.; Nathans, J. Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood-brain barrier and blood-retina barrier development and maintenance. Proc. Natl. Acad. Sci. USA 2018, 115, E11827–E11836. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Sapieha, P.; Sirinyan, M.; Hamel, D.; Zaniolo, K.; Joyal, J.S.; Cho, J.H.; Honore, J.C.; Kermorvant-Duchemin, E.; Varma, D.R.; Tremblay, S.; et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 2008, 14, 1067–1076. [Google Scholar] [CrossRef]
- Fulton, A.B.; Akula, J.D.; Mocko, J.A.; Hansen, R.M.; Benador, I.Y.; Beck, S.C.; Fahl, E.; Seeliger, M.W.; Moskowitz, A.; Harris, M.E. Retinal degenerative and hypoxic ischemic disease. Doc. Ophthalmol. Adv. Ophthalmol. 2009, 118, 55–61. [Google Scholar] [CrossRef]
- Akula, J.D.; Hansen, R.M.; Martinez-Perez, M.E.; Fulton, A.B. Rod photoreceptor function predicts blood vessel abnormality in retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4351–4359. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.M. Oxygen-induced retinopathy in mice with retinal photoreceptor cell degeneration. Life Sci. 2014, 102, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Gong, B.; Hatala, D.A.; Kern, T.S. Retinal ischemia and reperfusion causes capillary degeneration: Similarities to diabetes. Investig. Ophthalmol. Vis. Sci. 2007, 48, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakahara, T.; Hoshino, M.; Mori, A.; Sakamoto, K.; Ishii, K. Retinal blood vessels are damaged in a rat model of NMDA-induced retinal degeneration. Neurosci. Lett. 2010, 485, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Mori, A.; Kurauchi, Y.; Sakamoto, K.; Ishii, K. Neurovascular interactions in the retina: Physiological and pathological roles. J. Pharmacol. Sci. 2013, 123, 79–84. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, C.H.; Wang, Z.; Meng, S.S.; Burnim, S.B.; SanGiovanni, J.P.; Kamenecka, T.M.; Solt, L.A.; Chen, J. RORalpha modulates semaphorin 3E transcription and neurovascular interaction in pathological retinal angiogenesis. FASEB J. 2017, 31, 4492–4502. [Google Scholar] [CrossRef]
- Lange, C.A.; Bainbridge, J.W. Oxygen sensing in retinal health and disease. Ophthalmologica 2012, 227, 115–131. [Google Scholar] [CrossRef]
- Hoang, Q.V.; Linsenmeier, R.A.; Chung, C.K.; Curcio, C.A. Photoreceptor inner segments in monkey and human retina: Mitochondrial density, optics, and regional variation. Vis. Neurosci. 2002, 19, 395–407. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.J. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog. Retin. Eye Res. 2001, 20, 175–208. [Google Scholar] [CrossRef]
- Calzia, D.; Degan, P.; Caicci, F.; Bruschi, M.; Manni, L.; Ramenghi, L.A.; Candiano, G.; Traverso, C.E.; Panfoli, I. Modulation of the rod outer segment aerobic metabolism diminishes the production of radicals due to light absorption. Free Radic. Biol. Med. 2018, 117, 110–118. [Google Scholar] [CrossRef]
- Panfoli, I.; Calzia, D.; Bruschi, M.; Oneto, M.; Bianchini, P.; Ravera, S.; Petretto, A.; Diaspro, A.; Candiano, G. Functional expression of oxidative phosphorylation proteins in the rod outer segment disc. Cell Biochem. Funct. 2013, 31, 532–538. [Google Scholar] [CrossRef]
- Panfoli, I.; Calzia, D.; Bianchini, P.; Ravera, S.; Diaspro, A.; Candiano, G.; Bachi, A.; Monticone, M.; Aluigi, M.G.; Barabino, S.; et al. Evidence for aerobic metabolism in retinal rod outer segment disks. Int. J. Biochem. Cell Biol. 2009, 41, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Molday, R.S. Glucose metabolism in photoreceptor outer segments. Its role in phototransduction and in NADPH-requiring reactions. J. Biol. Chem. 1994, 269, 17954–17959. [Google Scholar] [PubMed]
- Stefansson, E.; Olafsdottir, O.B.; Einarsdottir, A.B.; Eliasdottir, T.S.; Eysteinsson, T.; Vehmeijer, W.; Vandewalle, E.; Bek, T.; Hardarson, S.H. Retinal Oximetry Discovers Novel Biomarkers in Retinal and Brain Diseases. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO227–BIO233. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Vascular responses to hypoxia and ischemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Rosenstein, J.M.; Krum, J.M.; Ruhrberg, C. VEGF in the nervous system. Organogenesis 2010, 6, 107–114. [Google Scholar] [CrossRef]
- Mackenzie, F.; Ruhrberg, C. Diverse roles for VEGF-A in the nervous system. Development 2012, 139, 1371–1380. [Google Scholar] [CrossRef]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Pierce, E.A.; Avery, R.L.; Foley, E.D.; Aiello, L.P.; Smith, L.E. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc. Natl. Acad. Sci. USA 1995, 92, 905–909. [Google Scholar] [CrossRef]
- Akula, J.D.; Hansen, R.M.; Tzekov, R.; Favazza, T.L.; Vyhovsky, T.C.; Benador, I.Y.; Mocko, J.A.; McGee, D.; Kubota, R.; Fulton, A.B. Visual cycle modulation in neurovascular retinopathy. Exp. Eye Res. 2010, 91, 153–161. [Google Scholar] [CrossRef]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.H.; Noell, W.K. Glucose catabolism of rabbit retina before and after development of visual function. J. Neurochem. 1960, 5, 253–276. [Google Scholar] [CrossRef]
- Dourlen, P.; Sujkowski, A.; Wessells, R.; Mollereau, B. Fatty acid transport proteins in disease: New insights from invertebrate models. Prog. Lipid Res. 2015, 60, 30–40. [Google Scholar] [CrossRef]
- Folz, S.J.; Trobe, J.D. The peroxisome and the eye. Surv. Ophthalmol. 1991, 35, 353–368. [Google Scholar] [CrossRef]
- Yagita, Y.; Shinohara, K.; Abe, Y.; Nakagawa, K.; Al-Owain, M.; Alkuraya, F.S.; Fujiki, Y. Deficiency of a Retinal Dystrophy Protein, Acyl-CoA Binding Domain-containing 5 (ACBD5), Impairs Peroxisomal beta-Oxidation of Very-long-chain Fatty Acids. J. Biol. Chem. 2017, 292, 691–705. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Falkenberg, K.D.; Koster, J.; Mooyer, P.A.; Jones, R.; van Roermund, C.W.T.; Pizzino, A.; Schrader, M.; Wanders, R.J.A.; Vanderver, A.; et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med. Genet. 2017, 54, 330–337. [Google Scholar] [CrossRef]
- Fu, Z.; Lofqvist, C.A.; Liegl, R.; Wang, Z.; Sun, Y.; Gong, Y.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Photoreceptor glucose metabolism determines normal retinal vascular growth. EMBO Mol. Med. 2018, 10, 79–90. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Ohue-Kitano, R.; Mukouyama, H.; Nishida, A.; Watanabe, K.; Igarashi, M.; Irie, J.; Tsujimoto, G.; Satoh-Asahara, N.; Itoh, H.; et al. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc. Natl. Acad. Sci. USA 2019, 116, 23813–23821. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Izuta, Y.; Imada, T.; Hisamura, R.; Oonishi, E.; Nakamura, S.; Inagaki, E.; Ito, M.; Soga, T.; Tsubota, K. Ketone body 3-hydroxybutyrate mimics calorie restriction via the Nrf2 activator, fumarate, in the retina. Aging Cell 2018, 17, e12699. [Google Scholar] [CrossRef] [PubMed]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurle, J.B.; Philp, N.J. The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem. 2014, 289, 20570–20582. [Google Scholar] [CrossRef]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef]
- Avogaro, A.; Crepaldi, C.; Miola, M.; Maran, A.; Pengo, V.; Tiengo, A.; Del Prato, S. High blood ketone body concentration in type 2 non-insulin dependent diabetic patients. J. Endocrinol. Investig. 1996, 19, 99–105. [Google Scholar] [CrossRef]
- Mahendran, Y.; Vangipurapu, J.; Cederberg, H.; Stancakova, A.; Pihlajamaki, J.; Soininen, P.; Kangas, A.J.; Paananen, J.; Civelek, M.; Saleem, N.K.; et al. Association of ketone body levels with hyperglycemia and type 2 diabetes in 9398 Finnish men. Diabetes 2013, 62, 3618–3626. [Google Scholar] [CrossRef]
- Harano, Y.; Kosugi, K.; Hyosu, T.; Suzuki, M.; Hidaka, H.; Kashiwagi, A.; Uno, S.; Shigeta, Y. Ketone bodies as markers for type 1 (insulin-dependent) diabetes and their value in the monitoring of diabetic control. Diabetologia 1984, 26, 343–348. [Google Scholar] [CrossRef]
- Fromal, O.; Lamoke, F.; Kaufman, M.; Bartoli, M.; Martin, P.M. Blockade of NLRP3 inflammasome activation in diabetic retina by the ketone metabolite beta-hydroxybutyrate is mediated by GPR109A. In ARVO Annual Meeting; ARVO: Seattle, WA, USA, 2016; Volume 57, p. 5451. [Google Scholar]
- Nishimura, Y.; Hara, H.; Kondo, M.; Hong, S.; Matsugi, T. Oxidative Stress in Retinal Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 4076518. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Lin, H.; Godley, B.F.; Boulton, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schumann, U.; Ader, M.; Brunssen, C.; Bramke, S.; Morawietz, H.; Funk, R.H. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; MacColl Garfinkel, A.E.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Zhu, B.T. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic. Biol. Med. 2010, 48, 821–830. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Calderon, G.D.; Juarez, O.H.; Hernandez, G.E.; Punzo, S.M.; De la Cruz, Z.D. Oxidative stress and diabetic retinopathy: Development and treatment. Eye (Lond.) 2017, 31, 1122–1130. [Google Scholar] [CrossRef]
- Li, S.Y.; Fu, Z.J.; Ma, H.; Jang, W.C.; So, K.F.; Wong, D.; Lo, A.C. Effect of lutein on retinal neurons and oxidative stress in a model of acute retinal ischemia/reperfusion. Investig. Ophthalmol. Vis. Sci. 2009, 50, 836–843. [Google Scholar] [CrossRef]
- Drobek-Slowik, M.; Karczewicz, D.; Safranow, K. The potential role of oxidative stress in the pathogenesis of the age-related macular degeneration (AMD). Postepy Hig. Med. Dosw. 2007, 61, 28–37. [Google Scholar]
- Ohashi, K.; Kageyama, M.; Shinomiya, K.; Fujita-Koyama, Y.; Hirai, S.I.; Katsuta, O.; Nakamura, M. Spermidine Oxidation-Mediated Degeneration of Retinal Pigment Epithelium in Rats. Oxid. Med. Cell. Longev. 2017, 2017, 4128061. [Google Scholar] [CrossRef] [PubMed]
- Cecilia, O.M.; Jose Alberto, C.G.; Jose, N.P.; Ernesto German, C.M.; Ana Karen, L.C.; Luis Miguel, R.P.; Ricardo Raul, R.R.; Adolfo Daniel, R.C. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J. Diabetes Res. 2019, 2019, 8562408. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, V.; Cherukuri, P.; Poria, D.; Goel, M.; Dagar, S.; Dhingra, N.K. Retinal Remodeling: Concerns, Emerging Remedies and Future Prospects. Front. Cell Neurosci. 2016, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kawasaki, R.; Dobson, L.P.; Ruddle, J.B.; Kearns, L.S.; Wong, T.Y.; Mackey, D.A. Quantitative analysis of retinal vessel attenuation in eyes with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Oishi, A.; Ogino, K.; Makiyama, Y.; Kurimoto, M.; Yoshimura, N. Association of retinal vessel attenuation with visual function in eyes with retinitis pigmentosa. Clin. Ophthalmol. 2014, 8, 1487–1493. [Google Scholar]
- Hanna, J.; Yucel, Y.H.; Zhou, X.; Mathieu, E.; Paczka-Giorgi, L.A.; Gupta, N. Progressive loss of retinal blood vessels in a live model of retinitis pigmentosa. Can. J. Ophthalmol. 2018, 53, 391–401. [Google Scholar] [CrossRef]
- Fernandez-Sanchez, L.; Esquiva, G.; Pinilla, I.; Lax, P.; Cuenca, N. Retinal Vascular Degeneration in the Transgenic P23H Rat Model of Retinitis Pigmentosa. Front. Neuroanat. 2018, 12, 55. [Google Scholar] [CrossRef]
- Dorrell, M.I.; Aguilar, E.; Jacobson, R.; Trauger, S.A.; Friedlander, J.; Siuzdak, G.; Friedlander, M. Maintaining retinal astrocytes normalizes revascularization and prevents vascular pathology associated with oxygen-induced retinopathy. Glia 2010, 58, 43–54. [Google Scholar] [CrossRef]
- Bucher, F.; Stahl, A.; Agostini, H.T.; Martin, G. Hyperoxia causes reduced density of retinal astrocytes in the central avascular zone in the mouse model of oxygen-induced retinopathy. Mol. Cell Neurosci. 2013, 56, 225–233. [Google Scholar] [CrossRef]
- O’Sullivan, M.L.; Punal, V.M.; Kerstein, P.C.; Brzezinski, J.A.T.; Glaser, T.; Wright, K.M.; Kay, J.N. Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia 2017, 65, 1697–1716. [Google Scholar] [CrossRef]
- Fu, Z.; Nian, S.; Li, S.Y.; Wong, D.; Chung, S.K.; Lo, A.C. Deficiency of aldose reductase attenuates inner retinal neuronal changes in a mouse model of retinopathy of prematurity. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 1503–1513. [Google Scholar] [CrossRef]
- Rubsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef]
- Rathi, S.; Jalali, S.; Patnaik, S.; Shahulhameed, S.; Musada, G.R.; Balakrishnan, D.; Rani, P.K.; Kekunnaya, R.; Chhablani, P.P.; Swain, S.; et al. Abnormal Complement Activation and Inflammation in the Pathogenesis of Retinopathy of Prematurity. Front. Immunol. 2017, 8, 1868. [Google Scholar] [CrossRef]
- Rivera, J.C.; Holm, M.; Austeng, D.; Morken, T.S.; Zhou, T.E.; Beaudry-Richard, A.; Sierra, E.M.; Dammann, O.; Chemtob, S. Retinopathy of prematurity: Inflammation, choroidal degeneration, and novel promising therapeutic strategies. J. Neuroinflam. 2017, 14, 165. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef]
- Mansoor, N.; Wahid, F.; Azam, M.; Shah, K.; den Hollander, A.I.; Qamar, R.; Ayub, H. Molecular Mechanisms of Complement System Proteins and Matrix Metalloproteinases in the Pathogenesis of Age-Related Macular Degeneration. Curr. Mol. Med. 2019, 19, 705–718. [Google Scholar] [CrossRef]
- Chen, M.; Citil, A.; McCabe, F.; Leicht, K.M.; Fiascone, J.; Dammann, C.E.; Dammann, O. Infection, oxygen, and immaturity: Interacting risk factors for retinopathy of prematurity. Neonatology 2011, 99, 125–132. [Google Scholar] [CrossRef]
- Stahl, A.; Joyal, J.S.; Chen, J.; Sapieha, P.; Juan, A.M.; Hatton, C.J.; Pei, D.T.; Hurst, C.G.; Seaward, M.R.; Krah, N.M.; et al. SOCS3 is an endogenous inhibitor of pathologic angiogenesis. Blood 2012, 120, 2925–2929. [Google Scholar] [CrossRef]
- Sun, Y.; Ju, M.; Lin, Z.; Fredrick, T.W.; Evans, L.P.; Tian, K.T.; Saba, N.J.; Morss, P.C.; Pu, W.T.; Chen, J.; et al. SOCS3 in retinal neurons and glial cells suppresses VEGF signaling to prevent pathological neovascular growth. Sci. Signal. 2015, 8, ra94. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, C.H.; SanGiovanni, J.P.; Evans, L.P.; Tian, K.T.; Zhang, B.; Stahl, A.; Pu, W.T.; Kamenecka, T.M.; Solt, L.A.; et al. Nuclear receptor RORalpha regulates pathologic retinal angiogenesis by modulating SOCS3-dependent inflammation. Proc. Natl. Acad. Sci. USA 2015, 112, 10401–10406. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- de Oliveira-Marques, V.; Cyrne, L.; Marinho, H.S.; Antunes, F. A quantitative study of NF-kappaB activation by H2O2: Relevance in inflammation and synergy with TNF-alpha. J. Immunol. 2007, 178, 3893–3902. [Google Scholar] [CrossRef]
- Volpe, J.J. Postnatal sepsis, necrotizing entercolitis, and the critical role of systemic inflammation in white matter injury in premature infants. J. Pediatr. 2008, 153, 160–163. [Google Scholar] [CrossRef]
- Silveira, R.C.; Fortes Filho, J.B.; Procianoy, R.S. Assessment of the contribution of cytokine plasma levels to detect retinopathy of prematurity in very low birth weight infants. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1297–1301. [Google Scholar] [CrossRef]
- Lavoie, P.M.; Lavoie, J.C.; Watson, C.; Rouleau, T.; Chang, B.A.; Chessex, P. Inflammatory response in preterm infants is induced early in life by oxygen and modulated by total parenteral nutrition. Pediatr. Res. 2010, 68, 248–251. [Google Scholar] [CrossRef]
- Dammann, O. Inflammation and retinopathy of prematurity. Acta Paediatr. 2010, 99, 975–977. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Gibson, D.S.; de Gooyer, T.E.; de la Cruz, V.F.; McDonald, D.M.; Stitt, A.W. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am. J. Pathol. 2005, 166, 637–644. [Google Scholar] [CrossRef]
- Connor, K.M.; SanGiovanni, J.P.; Lofqvist, C.; Aderman, C.M.; Chen, J.; Higuchi, A.; Hong, S.; Pravda, E.A.; Majchrzak, S.; Carper, D.; et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 2007, 13, 868–873. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, Z.; Liu, C.H.; Gong, Y.; Liegl, R.; Fredrick, T.W.; Meng, S.S.; Burnim, S.B.; Wang, Z.; Akula, J.D.; et al. Inflammatory signals from photoreceptor modulate pathological retinal angiogenesis via c-Fos. J. Exp. Med. 2017, 214, 1753–1767. [Google Scholar] [CrossRef]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef]
- Miller, J.W. Age-related macular degeneration revisited--piecing the puzzle: The LXIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 2013, 155, 1–35 e13. [Google Scholar] [CrossRef]
- Kumar, A.; Zhao, L.; Fariss, R.N.; McMenamin, P.G.; Wong, W.T. Vascular associations and dynamic process motility in perivascular myeloid cells of the mouse choroid: Implications for function and senescent change. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1787–1796. [Google Scholar] [CrossRef]
- Skeie, J.M.; Mullins, R.F. Macrophages in neovascular age-related macular degeneration: Friends or foes? Eye 2009, 23, 747–755. [Google Scholar] [CrossRef]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925. [Google Scholar] [CrossRef]
- Chen, M.; Lechner, J.; Zhao, J.; Toth, L.; Hogg, R.; Silvestri, G.; Kissenpfennig, A.; Chakravarthy, U.; Xu, H. STAT3 Activation in Circulating Monocytes Contributes to Neovascular Age-Related Macular Degeneration. Curr. Mol. Med. 2016, 16, 412–423. [Google Scholar] [CrossRef]
- Apte, R.S.; Richter, J.; Herndon, J.; Ferguson, T.A. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Med. 2006, 3, e310. [Google Scholar] [CrossRef]
- Yang, P.; de Vos, A.F.; Kijlstra, A. Macrophages and MHC class II positive cells in the choroid during endotoxin induced uveitis. Br. J. Ophthalmol. 1997, 81, 396–401. [Google Scholar] [CrossRef][Green Version]
- McMenamin, P.G. Dendritic cells and macrophages in the uveal tract of the normal mouse eye. Br. J. Ophthalmol. 1999, 83, 598–604. [Google Scholar] [CrossRef]
- Forrester, J.V.; Xu, H.; Kuffova, L.; Dick, A.D.; McMenamin, P.G. Dendritic cell physiology and function in the eye. Immunol. Rev. 2010, 234, 282–304. [Google Scholar] [CrossRef]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.; Allen, D.M.; Cardona, A.E. The role of microglia in diabetic retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Reichhart, N.; Hernandez-Matas, C.; Zabulis, X.; Kociok, N.; Brockmann, C.; Joussen, A.M.; Strauss, O. In vivo analysis of the time and spatial activation pattern of microglia in the retina following laser-induced choroidal neovascularization. Exp. Eye Res. 2015, 139, 13–21. [Google Scholar] [CrossRef]
- Talia, D.M.; Deliyanti, D.; Agrotis, A.; Wilkinson-Berka, J.L. Inhibition of the Nuclear Receptor RORgamma and Interleukin-17A Suppresses Neovascular Retinopathy: Involvement of Immunocompetent Microglia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1186–1196. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Schmid, M.C.; Varner, J.A. Myeloid cell trafficking and tumor angiogenesis. Cancer Lett. 2007, 250, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, T.A.; Apte, R.S. Angiogenesis in eye disease: Immunity gained or immunity lost? Semin. Immunopathol. 2008, 30, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Burris, T.P. Action of RORs and their ligands in (patho)physiology. Trends Endocrinol. Metab. 2012, 23, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009, 7, e003. [Google Scholar] [CrossRef]
- Houston, S.K.; Bourne, T.D.; Lopes, M.B.; Ghazi, N.G. Bilateral massive retinal gliosis associated with retinopathy of prematurity. Arch. Pathol. Lab. Med. 2009, 133, 1242–1245. [Google Scholar]
- Gu, L.; Xu, H.; Zhang, C.; Yang, Q.; Zhang, L.; Zhang, J. Time-dependent changes in hypoxia- and gliosis-related factors in experimental diabetic retinopathy. Eye 2019, 33, 600–609. [Google Scholar] [CrossRef]
- Kern, T.S. Interrelationships between the Retinal Neuroglia and Vasculature in Diabetes. Diabetes Metab. J. 2014, 38, 163–170. [Google Scholar] [CrossRef]
- Qian, H.; Ripps, H. Neurovascular interaction and the pathophysiology of diabetic retinopathy. Exp. Diabetes Res. 2011, 2011, 693426. [Google Scholar] [CrossRef]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Nishijima, T.; Piriz, J.; Duflot, S.; Fernandez, A.M.; Gaitan, G.; Gomez-Pinedo, U.; Verdugo, J.M.; Leroy, F.; Soya, H.; Nunez, A.; et al. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 2010, 67, 834–846. [Google Scholar] [CrossRef]
- Luo, Y.; Raible, D.; Raper, J.A. Collapsin: A protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 1993, 75, 217–227. [Google Scholar] [CrossRef]
- Gaur, P.; Bielenberg, D.R.; Samuel, S.; Bose, D.; Zhou, Y.; Gray, M.J.; Dallas, N.A.; Fan, F.; Xia, L.; Lu, J.; et al. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin. Cancer Res. 2009, 15, 6763–6770. [Google Scholar] [CrossRef]
- Alto, L.T.; Terman, J.R. Semaphorins and their Signaling Mechanisms. Methods Mol. Biol. 2017, 1493, 1–25. [Google Scholar]
- Toledano, S.; Nir-Zvi, I.; Engelman, R.; Kessler, O.; Neufeld, G. Class-3 Semaphorins and Their Receptors: Potent Multifunctional Modulators of Tumor Progression. Int. J. Mol. Sci. 2019, 20, 556. [Google Scholar] [CrossRef]
- Franzolin, G.; Tamagnone, L. Semaphorin Signaling in Cancer-Associated Inflammation. Int. J. Mol. Sci. 2019, 20, 377. [Google Scholar] [CrossRef]
- Cerani, A.; Tetreault, N.; Menard, C.; Lapalme, E.; Patel, C.; Sitaras, N.; Beaudoin, F.; Leboeuf, D.; De Guire, V.; Binet, F.; et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab. 2013, 18, 505–518. [Google Scholar] [CrossRef]
- Yang, W.J.; Hu, J.; Uemura, A.; Tetzlaff, F.; Augustin, H.G.; Fischer, A. Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med. 2015, 7, 1267–1284. [Google Scholar] [CrossRef]
- Wu, J.H.; Li, Y.N.; Chen, A.Q.; Hong, C.D.; Zhang, C.L.; Wang, H.L.; Zhou, Y.F.; Li, P.C.; Wang, Y.; Mao, L.; et al. Inhibition of Sema4D/PlexinB1 signaling alleviates vascular dysfunction in diabetic retinopathy. EMBO Mol. Med. 2020, 12, e10154. [Google Scholar] [CrossRef]
- Fukushima, Y.; Okada, M.; Kataoka, H.; Hirashima, M.; Yoshida, Y.; Mann, F.; Gomi, F.; Nishida, K.; Nishikawa, S.; Uemura, A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J. Clin. Investig. 2011, 121, 1974–1985. [Google Scholar] [CrossRef] [PubMed]
- Klufas, M.A.; Chan, R.V. Intravitreal anti-VEGF therapy as a treatment for retinopathy of prematurity: What we know after 7 years. J. Pediatr. Ophthalmol. Strabismus 2015, 52, 77–84. [Google Scholar] [CrossRef]
- Bakri, S.J.; Thorne, J.E.; Ho, A.C.; Ehlers, J.P.; Schoenberger, S.D.; Yeh, S.; Kim, S.J. Safety and Efficacy of Anti-Vascular Endothelial Growth Factor Therapies for Neovascular Age-Related Macular Degeneration: A Report by the American Academy of Ophthalmology. Ophthalmology 2019, 126, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nigam, N.; Hedaya, J.; Freeman, W.R. Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. Br. J. Ophthalmol. 2008, 92, 865–866. [Google Scholar]
- Lux, A.; Llacer, H.; Heussen, F.M.; Joussen, A.M. Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. Br. J. Ophthalmol. 2007, 91, 1318–1322. [Google Scholar] [CrossRef]
- Ventrice, P.; Leporini, C.; Aloe, J.F.; Greco, E.; Leuzzi, G.; Marrazzo, G.; Scorcia, G.B.; Bruzzichesi, D.; Nicola, V.; Scorcia, V. Anti-vascular endothelial growth factor drugs safety and efficacy in ophthalmic diseases. J. Pharm. Pharm. 2013, 4, S38–S42. [Google Scholar]
- Avery, R.L.; Castellarin, A.A.; Steinle, N.C.; Dhoot, D.S.; Pieramici, D.J.; See, R.; Couvillion, S.; Nasir, M.A.; Rabena, M.D.; Le, K.; et al. Systemic pharmacokinetics following intravitreal injections of ranibizumab, bevacizumab or aflibercept in patients with neovascular AMD. Br. J. Ophthalmol. 2014, 98, 1636–1641. [Google Scholar] [CrossRef]
- Bressler, N.M.; Boyer, D.S.; Williams, D.F.; Butler, S.; Francom, S.F.; Brown, B.; Di Nucci, F.; Cramm, T.; Tuomi, L.L.; Ianchulev, T.; et al. Cerebrovascular accidents in patients treated for choroidal neovascularization with ranibizumab in randomized controlled trials. Retina 2012, 32, 1821–1828. [Google Scholar] [CrossRef]
- Moorthy, S.; Cheung, N. Cerebrovascular accidents and ranibizumab. Ophthalmology 2009, 116, 1835. [Google Scholar] [CrossRef]
- Ueta, T.; Yanagi, Y.; Tamaki, Y.; Yamaguchi, T. Cerebrovascular accidents in ranibizumab. Ophthalmology 2009, 116, 362. [Google Scholar] [CrossRef]
- Hapani, S.; Sher, A.; Chu, D.; Wu, S. Increased risk of serious hemorrhage with bevacizumab in cancer patients: A meta-analysis. Oncology 2010, 79, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Balakrishnan, D.; Zeynalova, Z.; Padhi, T.R.; Rani, P.K. Serious adverse events and visual outcomes of rescue therapy using adjunct bevacizumab to laser and surgery for retinopathy of prematurity. The Indian Twin Cities Retinopathy of Prematurity Screening database Report number 5. Arch. Dis. Child. Fetal Neonatal Ed. 2013, 98, F327–F333. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, K.; Cooper, N.G. MicroRNAs in the Neural Retina. Int. J. Genom. 2014, 2014, 165897. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Chaqour, B. MicroRNA signature and function in retinal neovascularization. World J. Biol. Chem. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Wang, Z.; Huang, S.; Sun, Y.; Chen, J. MicroRNA-145 Regulates Pathological Retinal Angiogenesis by Suppression of TMOD3. Mol. Nucleic Acids 2019, 16, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Wang, Z.; Sun, Y.; SanGiovanni, J.P.; Chen, J. Retinal expression of small non-coding RNAs in a murine model of proliferative retinopathy. Sci. Rep. 2016, 6, 33947. [Google Scholar] [CrossRef]
- Liu, C.H.; Sun, Y.; Li, J.; Gong, Y.; Tian, K.T.; Evans, L.P.; Morss, P.C.; Fredrick, T.W.; Saba, N.J.; Chen, J. Endothelial microRNA-150 is an intrinsic suppressor of pathologic ocular neovascularization. Proc. Natl. Acad. Sci. USA 2015, 112, 12163–12168. [Google Scholar] [CrossRef]
- Walsky, R.L.; Gaman, E.A.; Obach, R.S. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J. Clin. Pharm. 2005, 45, 68–78. [Google Scholar] [CrossRef]
- Keech, A.C.; Mitchell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- Group, A.S.; Group, A.E.S.; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; Esser, B.A.; et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233–244. [Google Scholar]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, A.C.; Mott, R.; Lyons, T.J.; Ma, J.X. Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013, 62, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Shao, Z.; Fu, Z.; Edin, M.L.; Sun, Y.; Liegl, R.G.; Wang, Z.; Liu, C.H.; Burnim, S.B.; Meng, S.S.; et al. Fenofibrate Inhibits Cytochrome P450 Epoxygenase 2C Activity to Suppress Pathological Ocular Angiogenesis. EBioMedicine 2016, 13, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fu, Z.; Edin, M.L.; Liu, C.H.; Wang, Z.; Shao, Z.; Fredrick, T.W.; Saba, N.J.; Morss, P.C.; Burnim, S.B.; et al. Cytochrome P450 Oxidase 2C Inhibition Adds to omega-3 Long-Chain Polyunsaturated Fatty Acids Protection Against Retinal and Choroidal Neovascularization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Ding, L.; He, X.; Takahashi, Y.; Ma, J.X. Interaction of PPARalpha With the Canonic Wnt Pathway in the Regulation of Renal Fibrosis. Diabetes 2016, 65, 3730–3743. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular development in the retina and inner ear: Control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.; Ma, J.X. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. Physiol. 2016, 311, H738–H749. [Google Scholar] [CrossRef]
- Chen, J.; Stahl, A.; Krah, N.M.; Seaward, M.R.; Dennison, R.J.; Sapieha, P.; Hua, J.; Hatton, C.J.; Juan, A.M.; Aderman, C.M.; et al. Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation 2011, 124, 1871–1881. [Google Scholar] [CrossRef]
- Lundasen, T.; Hunt, M.C.; Nilsson, L.M.; Sanyal, S.; Angelin, B.; Alexson, S.E.; Rudling, M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Y.; Xiong, R.; Zheng, J.; Li, Q.; Zhang, S.; Li, X.; Pan, X.; Yang, S. FGF21 mediates the protective effect of fenofibrate against acetaminophen -induced hepatotoxicity via activating autophagy in mice. Biochem. Biophys. Res. Commun. 2018, 503, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.H.; Kim, H.K.; Kim, M.J.; Back, S.H.; Konishi, M.; Itoh, N.; Lee, M.S. Fibroblast growth factor 21 participates in adaptation to endoplasmic reticulum stress and attenuates obesity-induced hepatic metabolic stress. Diabetologia 2015, 58, 809–818. [Google Scholar] [CrossRef]
- Berglund, E.D.; Li, C.Y.; Bina, H.A.; Lynes, S.E.; Michael, M.D.; Shanafelt, A.B.; Kharitonenkov, A.; Wasserman, D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 2009, 150, 4084–4093. [Google Scholar] [CrossRef]
- Lin, Z.; Tian, H.; Lam, K.S.; Lin, S.; Hoo, R.C.; Konishi, M.; Itoh, N.; Wang, Y.; Bornstein, S.R.; Xu, A.; et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013, 17, 779–789. [Google Scholar] [CrossRef]
- Kim, H.W.; Lee, J.E.; Cha, J.J.; Hyun, Y.Y.; Kim, J.E.; Lee, M.H.; Song, H.K.; Nam, D.H.; Han, J.Y.; Han, S.Y.; et al. Fibroblast growth factor 21 improves insulin resistance and ameliorates renal injury in db/db mice. Endocrinology 2013, 154, 3366–3376. [Google Scholar] [CrossRef]
- Zhang, C.; Shao, M.; Yang, H.; Chen, L.; Yu, L.; Cong, W.; Tian, H.; Zhang, F.; Cheng, P.; Jin, L.; et al. Attenuation of hyperlipidemia- and diabetes-induced early-stage apoptosis and late-stage renal dysfunction via administration of fibroblast growth factor-21 is associated with suppression of renal inflammation. PLoS ONE 2013, 8, e82275. [Google Scholar] [CrossRef]
- Liu, J.J.; Foo, J.P.; Liu, S.; Lim, S.C. The role of fibroblast growth factor 21 in diabetes and its complications: A review from clinical perspective. Diabetes Res. Clin. Pract. 2015, 108, 382–389. [Google Scholar] [CrossRef]
- Schlein, C.; Talukdar, S.; Heine, M.; Fischer, A.W.; Krott, L.M.; Nilsson, S.K.; Brenner, M.B.; Heeren, J.; Scheja, L. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016, 23, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Lu, W.; Wang, X.; Wang, C.; Abbruzzese, J.L.; Liang, G.; Li, X.; Luo, Y. FGF21-FGFR1 Coordinates Phospholipid Homeostasis, Lipid Droplet Function, and ER Stress in Obesity. Endocrinology 2016, 157, 4754–4769. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Zhou, Y.; Li, D.; Rossulek, M.; Dong, J.; Somayaji, V.; Weng, Y.; Clark, R.; Lanba, A.; Owen, B.M.; et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016, 23, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef]
- Fu, Z.; Gong, Y.; Liegl, R.; Wang, Z.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Saba, N.J.; Fredrick, T.W.; Morss, P.C.; et al. FGF21 Administration Suppresses Retinal and Choroidal Neovascularization in Mice. Cell Rep. 2017, 18, 1606–1613. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, Z.; Liu, C.H.; Gong, Y.; Cakir, B.; Liegl, R.; Sun, Y.; Meng, S.; Burnim, S.; Arellano, I.; et al. Fibroblast growth factor 21 protects photoreceptor function in type 1 diabetic mice. Diabetes 2018, 67, 974–985. [Google Scholar] [CrossRef]
- Yu, Z.; Lin, L.; Jiang, Y.; Chin, I.; Wang, X.; Li, X.; Lo, E.H.; Wang, X. Recombinant FGF21 Protects Against Blood-Brain Barrier Leakage Through Nrf2 Upregulation in Type 2 Diabetes Mice. Mol. Neurobiol. 2019, 56, 2314–2327. [Google Scholar] [CrossRef]
- Li, H.; Wu, G.; Fang, Q.; Zhang, M.; Hui, X.; Sheng, B.; Wu, L.; Bao, Y.; Li, P.; Xu, A.; et al. Fibroblast growth factor 21 increases insulin sensitivity through specific expansion of subcutaneous fat. Nat. Commun. 2018, 9, 272. [Google Scholar] [CrossRef]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef]
- Fu, Z.; Gong, Y.; Lofqvist, C.; Hellstrom, A.; Smith, L.E. Review: Adiponectin in retinopathy. Biochim. Biophys. Acta 2016, 1862, 1392–1400. [Google Scholar] [CrossRef]
- Fu, Z.; Lofqvist, C.A.; Shao, Z.; Sun, Y.; Joyal, J.S.; Hurst, C.G.; Cui, R.Z.; Evans, L.P.; Tian, K.; SanGiovanni, J.P.; et al. Dietary omega-3 polyunsaturated fatty acids decrease retinal neovascularization by adipose-endoplasmic reticulum stress reduction to increase adiponectin. Am. J. Clin. Nutr. 2015, 101, 879–888. [Google Scholar] [CrossRef]
- Mao, D.; Peng, H.; Li, Q.; Wang, J.; Li, P.; Hu, K.; Zhang, X.; Lei, B. Aqueous humor and plasma adiponectin levels in proliferative diabetic retinopathy patients. Curr. Eye Res. 2012, 37, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Omae, T.; Nagaoka, T.; Yoshida, A. Relationship Between Retinal Blood Flow and Serum Adiponectin Concentrations in Patients With Type 2 Diabetes Mellitus. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4143–4149. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Paananen, J.; Nevalainen, T.; Sorri, I.; Seitsonen, S.; Immonen, I.; Salminen, A.; Pulkkinen, L.; Uusitupa, M. Adiponectin receptor 1 gene (ADIPOR1) variant is associated with advanced age-related macular degeneration in Finnish population. Neurosci. Lett. 2012, 513, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, C.; Shen, Y.; Chen, N.; Wang, L.; Liang, L.; Guo, T.; Yin, X.; Ma, Z.; Zhang, B.; et al. A mutation in ADIPOR1 causes nonsyndromic autosomal dominant retinitis pigmentosa. Hum. Genet. 2016, 135, 1375–1387. [Google Scholar] [CrossRef]
- Higuchi, A.; Ohashi, K.; Kihara, S.; Walsh, K.; Ouchi, N. Adiponectin suppresses pathological microvessel formation in retina through modulation of tumor necrosis factor-alpha expression. Circ. Res. 2009, 104, 1058–1065. [Google Scholar] [CrossRef]
- Lyzogubov, V.V.; Tytarenko, R.G.; Bora, N.S.; Bora, P.S. Inhibitory role of adiponectin peptide I on rat choroidal neovascularization. Biochim. Biophys. Acta 2012, 1823, 1264–1272. [Google Scholar] [CrossRef]
- Fu, Z.; Liegl, R.; Wang, Z.; Gong, Y.; Liu, C.H.; Sun, Y.; Cakir, B.; Burnim, S.B.; Meng, S.S.; Lofqvist, C.; et al. Adiponectin Mediates Dietary Omega-3 Long-Chain Polyunsaturated Fatty Acid Protection Against Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3862–3870. [Google Scholar] [CrossRef]
- Palanisamy, K.; Nareshkumar, R.N.; Sivagurunathan, S.; Raman, R.; Sulochana, K.N.; Chidambaram, S. Anti-angiogenic effect of adiponectin in human primary microvascular and macrovascular endothelial cells. Microvasc. Res. 2019, 122, 136–145. [Google Scholar] [CrossRef]
- Omae, T.; Nagaoka, T.; Tanano, I.; Yoshida, A. Adiponectin-induced dilation of isolated porcine retinal arterioles via production of nitric oxide from endothelial cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4586–4594. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Kamoshita, M.; Ozawa, Y.; Kubota, S.; Miyake, S.; Tsuda, C.; Nagai, N.; Yuki, K.; Shimmura, S.; Umezawa, K.; Tsubota, K. AMPK-NF-kappaB axis in the photoreceptor disorder during retinal inflammation. PLoS ONE 2014, 9, e103013. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kong, L.; Wang, J.; Ash, J.D. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2018, 115, 10475–10480. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1alpha Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1alpha pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1alpha repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model. Mech. 2018, 11, dmm032698. [Google Scholar] [CrossRef]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1alpha Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1alpha in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Egger, A.; Samardzija, M.; Sothilingam, V.; Tanimoto, N.; Lange, C.; Salatino, S.; Fang, L.; Garcia-Garrido, M.; Beck, S.; Okoniewski, M.J.; et al. PGC-1alpha determines light damage susceptibility of the murine retina. PLoS ONE 2012, 7, e31272. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Jiang, A.; Abend, S.; Liu, L.; Sweigard, H.; Connor, K.M.; Arany, Z. PGC-1alpha regulates normal and pathological angiogenesis in the retina. Am. J. Pathol. 2013, 182, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Deepa, S.S.; Etzler, J.C.; Ryu, J.; Mao, X.; Fang, Q.; Liu, D.D.; Torres, J.M.; Jia, W.; Lechleiter, J.D.; et al. Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J. Biol. Chem. 2009, 284, 22426–22435. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. FGF21 activates AMPK signaling: Impact on metabolic regulation and the aging process. J. Mol. Med. 2017, 95, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Peplow, P.V.; Chung, T.Y.; Baxter, G.D. Laser photobiomodulation of wound healing: A review of experimental studies in mouse and rat animal models. Photomed. Laser Surg. 2010, 28, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.T.; Johnson, M.I.; Lopes-Martins, R.A.; Bjordal, J.M. Efficacy of low-level laser therapy in the management of neck pain: A systematic review and meta-analysis of randomised placebo or active-treatment controlled trials. Lancet 2009, 374, 1897–1908. [Google Scholar] [CrossRef]
- Anders, J.J.; Geuna, S.; Rochkind, S. Phototherapy promotes regeneration and functional recovery of injured peripheral nerve. Neurol. Res. 2004, 26, 233–239. [Google Scholar] [CrossRef]
- Lampl, Y.; Zivin, J.A.; Fisher, M.; Lew, R.; Welin, L.; Dahlof, B.; Borenstein, P.; Andersson, B.; Perez, J.; Caparo, C.; et al. Infrared laser therapy for ischemic stroke: A new treatment strategy: Results of the NeuroThera Effectiveness and Safety Trial-1 (NEST-1). Stroke 2007, 38, 1843–1849. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Hodgetts, S.; Van Den Heuvel, C.; Natoli, R.; Hart, N.S.; Valter, K.; Harvey, A.R.; Vink, R.; Provis, J.; Dunlop, S.A. Red/near-infrared irradiation therapy for treatment of central nervous system injuries and disorders. Rev. Neurosci. 2013, 24, 205–226. [Google Scholar] [CrossRef]
- Rutar, M.; Natoli, R.; Albarracin, R.; Valter, K.; Provis, J. 670-nm light treatment reduces complement propagation following retinal degeneration. J. Neuroinflam. 2012, 9, 257. [Google Scholar] [CrossRef]
- Albarracin, R.; Eells, J.; Valter, K. Photobiomodulation protects the retina from light-induced photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3582–3592. [Google Scholar] [CrossRef] [PubMed]
- Albarracin, R.; Natoli, R.; Rutar, M.; Valter, K.; Provis, J. 670 nm light mitigates oxygen-induced degeneration in C57BL/6J mouse retina. BMC Neurosci. 2013, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Natoli, R.; Valter, K.; Barbosa, M.; Dahlstrom, J.; Rutar, M.; Kent, A.; Provis, J. 670 nm photobiomodulation as a novel protection against retinopathy of prematurity: Evidence from oxygen induced retinopathy models. PLoS ONE 2013, 8, e72135. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Du, Y.; Lee, C.A.; Talahalli, R.; Eells, J.T.; Kern, T.S. Low-intensity far-red light inhibits early lesions that contribute to diabetic retinopathy: In vivo and in vitro. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3681–3690. [Google Scholar] [CrossRef] [PubMed]
- Eells, J.T.; Henry, M.M.; Summerfelt, P.; Wong-Riley, M.T.; Buchmann, E.V.; Kane, M.; Whelan, N.T.; Whelan, H.T. Therapeutic photobiomodulation for methanol-induced retinal toxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3439–3444. [Google Scholar] [CrossRef]
- Begum, R.; Powner, M.B.; Hudson, N.; Hogg, C.; Jeffery, G. Treatment with 670 nm light up regulates cytochrome C oxidase expression and reduces inflammation in an age-related macular degeneration model. PLoS ONE 2013, 8, e57828. [Google Scholar] [CrossRef]
- Ivandic, B.T.; Ivandic, T. Low-level laser therapy improves vision in patients with age-related macular degeneration. Photomed. Laser Surg. 2008, 26, 241–245. [Google Scholar] [CrossRef]
- Merry, G.F.; Munk, M.R.; Dotson, R.S.; Walker, M.G.; Devenyi, R.G. Photobiomodulation reduces drusen volume and improves visual acuity and contrast sensitivity in dry age-related macular degeneration. Acta Ophthalmol. 2017, 95, e270–e277. [Google Scholar] [CrossRef]
- Gkotsi, D.; Begum, R.; Salt, T.; Lascaratos, G.; Hogg, C.; Chau, K.Y.; Schapira, A.H.; Jeffery, G. Recharging mitochondrial batteries in old eyes. Near infra-red increases ATP. Exp. Eye Res. 2014, 122, 50–53. [Google Scholar] [CrossRef]
- Kokkinopoulos, I.; Colman, A.; Hogg, C.; Heckenlively, J.; Jeffery, G. Age-related retinal inflammation is reduced by 670 nm light via increased mitochondrial membrane potential. Neurobiol. Aging 2013, 34, 602–609. [Google Scholar] [CrossRef]
- Karu, T.I. Mitochondrial signaling in mammalian cells activated by red and near-IR radiation. Photochem. Photobiol. 2008, 84, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Kaynezhad, P.; Tachtsidis, I.; Jeffery, G. Optical monitoring of retinal respiration in real time: 670 nm light increases the redox state of mitochondria. Exp. Eye Res. 2016, 152, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Beirne, K.; Rozanowska, M.; Votruba, M. Photostimulation of mitochondria as a treatment for retinal neurodegeneration. Mitochondrion 2017, 36, 85–95. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, C.T.; Cagnone, G.; Heckel, E.; Sun, Y.; Cakir, B.; Tomita, Y.; Huang, S.; Li, Q.; Britton, W.; et al. Dyslipidemia in retinal metabolic disorders. EMBO Mol. Med. 2019, 11, e10473. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tang, J.; Du, Y.; Saadane, A.; Samuels, I.; Veenstra, A.; Kiser, J.Z.; Palczewski, K.; Kern, T.S. Transducin1, Phototransduction and the Development of Early Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tang, J.; Du, Y.; Lee, C.A.; Golczak, M.; Muthusamy, A.; Antonetti, D.A.; Veenstra, A.A.; Amengual, J.; von Lintig, J.; et al. Retinylamine Benefits Early Diabetic Retinopathy in Mice. J. Biol. Chem. 2015, 290, 21568–21579. [Google Scholar] [CrossRef]
- Bavik, C.; Henry, S.H.; Zhang, Y.; Mitts, K.; McGinn, T.; Budzynski, E.; Pashko, A.; Lieu, K.L.; Zhong, S.; Blumberg, B.; et al. Visual Cycle Modulation as an Approach toward Preservation of Retinal Integrity. PLoS ONE 2015, 10, e0124940. [Google Scholar] [CrossRef]
- Tonade, D.; Liu, H.; Palczewski, K.; Kern, T.S. Photoreceptor cells produce inflammatory products that contribute to retinal vascular permeability in a mouse model of diabetes. Diabetologia 2017, 60, 2111–2120. [Google Scholar] [CrossRef]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef]
- Liu, H.; Tang, J.; Du, Y.; Saadane, A.; Tonade, D.; Samuels, I.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor Cells Influence Retinal Vascular Degeneration in Mouse Models of Retinal Degeneration and Diabetes. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4272–4281. [Google Scholar] [CrossRef]
- Kern, T.S.; Berkowitz, B.A. Photoreceptors in diabetic retinopathy. J. Diabetes Investig. 2015, 6, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Curran, T.; Franza, B.R., Jr. Fos and Jun: The AP-1 connection. Cell 1988, 55, 395–397. [Google Scholar] [CrossRef]
- Hafezi, F.; Steinbach, J.P.; Marti, A.; Munz, K.; Wang, Z.Q.; Wagner, E.F.; Aguzzi, A.; Reme, C.E. The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat. Med. 1997, 3, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.E.; Smith, M.S.; Verbalis, J.G. c-Fos and related immediate early gene products as markers of activity in neuroendocrine systems. Front. Neuroendocr. 1993, 14, 173–213. [Google Scholar] [CrossRef]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef]
- Shiozawa, S.; Tsumiyama, K. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle 2009, 8, 1539–1543. [Google Scholar] [CrossRef]
- Yu, M.C.; Li, W.W.; Liu, K.; Yew, D.T. An immunohistochemical study of the c-fos protooncogene in the developing human retina. Neuroscience 1994, 60, 983–987. [Google Scholar] [CrossRef]
- He, L.; Campbell, M.L.; Srivastava, D.; Blocker, Y.S.; Harris, J.R.; Swaroop, A.; Fox, D.A. Spatial and temporal expression of AP-1 responsive rod photoreceptor genes and bZIP transcription factors during development of the rat retina. Mol. Vis. 1998, 4, 32. [Google Scholar]
- Hobson, A.H.; Donovan, M.; Humphries, M.M.; Tuohy, G.; McNally, N.; Carmody, R.; Cotter, T.; Farrar, G.J.; Kenna, P.F.; Humphries, P. Apoptotic photoreceptor death in the rhodopsin knockout mouse in the presence and absence of c-fos. Exp. Eye Res. 2000, 71, 247–254. [Google Scholar] [CrossRef]
- Poon, H.K.; Tso, M.O.; Lam, T.T. c-Fos protein in photoreceptor cell death after photic injury in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2755–2758. [Google Scholar]
- Wenzel, A.; Grimm, C.; Marti, A.; Kueng-Hitz, N.; Hafezi, F.; Niemeyer, G.; Reme, C.E. c-fos controls the "private pathway" of light-induced apoptosis of retinal photoreceptors. J. Neurosci. 2000, 20, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Nathaniel, T.I.; Otukonyong, E.; Abdellatif, A.; Soyinka, J.O. Effect of hypoxia on metabolic rate, core body temperature, and c-fos expression in the naked mole rat. Int. J. Dev. Neurosci. 2012, 30, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Nakao, K.; Shimazaki, T.; Shimmura, S.; Kurihara, T.; Ishida, S.; Yoshimura, A.; Tsubota, K.; Okano, H. SOCS3 is required to temporally fine-tune photoreceptor cell differentiation. Dev. Biol. 2007, 303, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Offermanns, S. Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov. 2014, 13, 603–621. [Google Scholar] [CrossRef]
- Kim, J.; Oh, W.J.; Gaiano, N.; Yoshida, Y.; Gu, C. Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011, 25, 1399–1411. [Google Scholar] [CrossRef]
- Buehler, A.; Sitaras, N.; Favret, S.; Bucher, F.; Berger, S.; Pielen, A.; Joyal, J.S.; Juan, A.M.; Martin, G.; Schlunck, G.; et al. Semaphorin 3F forms an anti-angiogenic barrier in outer retina. FEBS Lett. 2013, 587, 1650–1655. [Google Scholar] [CrossRef]
- Ochsenbein, A.M.; Karaman, S.; Proulx, S.T.; Berchtold, M.; Jurisic, G.; Stoeckli, E.T.; Detmar, M. Endothelial cell-derived semaphorin 3A inhibits filopodia formation by blood vascular tip cells. Development 2016, 143, 589–594. [Google Scholar] [CrossRef]
- Joyal, J.S.; Sitaras, N.; Binet, F.; Rivera, J.C.; Stahl, A.; Zaniolo, K.; Shao, Z.; Polosa, A.; Zhu, T.; Hamel, D.; et al. Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A. Blood 2011, 117, 6024–6035. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Sun, Y.; Liegl, R.; Gong, Y.; Buhler, A.; Cakir, B.; Meng, S.S.; Burnim, S.B.; Liu, C.H.; Reuer, T.; Zhang, P.; et al. Sema3f Protects Against Subretinal Neovascularization In Vivo. EBioMedicine 2017, 18, 281–287. [Google Scholar] [CrossRef]
- Ajlan, R.S.; Silva, P.S.; Sun, J.K. Vascular Endothelial Growth Factor and Diabetic Retinal Disease. Semin. Ophthalmol. 2016, 31, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Choudhary, M.M.; Schachat, A.P. Neovascular Age-Related Macular Degeneration. Dev. Ophthalmol. 2016, 55, 125–136. [Google Scholar] [PubMed]
- Meyer, C.H.; Krohne, T.U.; Charbel Issa, P.; Liu, Z.; Holz, F.G. Routes for Drug Delivery to the Eye and Retina: Intravitreal Injections. Dev. Ophthalmol. 2016, 55, 63–70. [Google Scholar] [PubMed]
- Jaffe, G.J.; Eliott, D.; Wells, J.A.; Prenner, J.L.; Papp, A.; Patel, S. A Phase 1 Study of Intravitreous E10030 in Combination with Ranibizumab in Neovascular Age-Related Macular Degeneration. Ophthalmology 2016, 123, 78–85. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Z.; Sun, Y.; Cakir, B.; Tomita, Y.; Huang, S.; Wang, Z.; Liu, C.-H.; S. Cho, S.; Britton, W.; S. Kern, T.; et al. Targeting Neurovascular Interaction in Retinal Disorders. Int. J. Mol. Sci. 2020, 21, 1503. https://doi.org/10.3390/ijms21041503
Fu Z, Sun Y, Cakir B, Tomita Y, Huang S, Wang Z, Liu C-H, S. Cho S, Britton W, S. Kern T, et al. Targeting Neurovascular Interaction in Retinal Disorders. International Journal of Molecular Sciences. 2020; 21(4):1503. https://doi.org/10.3390/ijms21041503
Chicago/Turabian StyleFu, Zhongjie, Ye Sun, Bertan Cakir, Yohei Tomita, Shuo Huang, Zhongxiao Wang, Chi-Hsiu Liu, Steve S. Cho, William Britton, Timothy S. Kern, and et al. 2020. "Targeting Neurovascular Interaction in Retinal Disorders" International Journal of Molecular Sciences 21, no. 4: 1503. https://doi.org/10.3390/ijms21041503
APA StyleFu, Z., Sun, Y., Cakir, B., Tomita, Y., Huang, S., Wang, Z., Liu, C.-H., S. Cho, S., Britton, W., S. Kern, T., Antonetti, D. A., Hellström, A., & E.H. Smith, L. (2020). Targeting Neurovascular Interaction in Retinal Disorders. International Journal of Molecular Sciences, 21(4), 1503. https://doi.org/10.3390/ijms21041503